Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
11. Molekuladinamika és dokkolás.
Bioinformatikai programcsomagok.
Bioinformatika a gyakorlatban
Molekuladinamika
A fehérje mozgásainak szimulációja
A potenciálisenergia-felület
- Born-Oppenheimer-közelítés: az elektronok mozgása sokkal gyorsabb az
atommagokénál, az atommagok nehezek
- Ezért a molekuláris mozgások modellezésénél a molekulát klasszikus
rendszernek tekinthetjük: rugókkal összekötött, töltéssel rendelkező
tömegpontok.
- Az atommagok pozícióitól függő E(R) potenciálfüggvényt empirikus energiafüggvénnyel
helyettesíthetjük (a Schrödinger-egyenlet megoldása helyett)
- Az atommagok mozgásának leírására
használhatjuk a Newton-féle mozgásegyenletet:
-(dE/dR) =
m(d2R/dt2)
Ez a molekuladinamika.
A forcefield
- A forcefield az empirikus energiafüggvény matematikai alakja. Egy
tipikus forcefield:
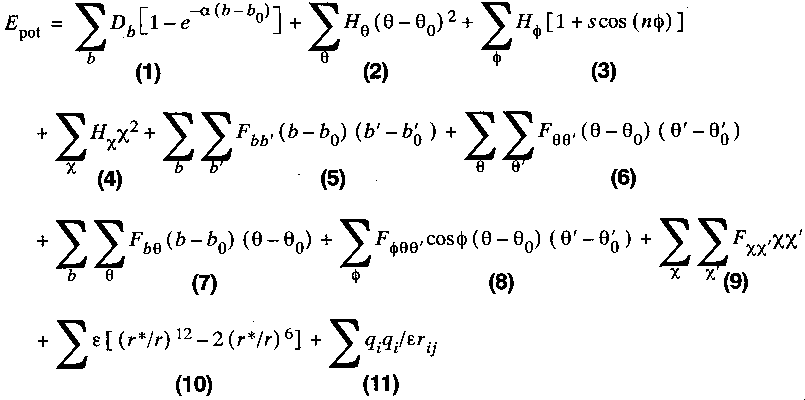 Az egyes tagok jelentése:
Az egyes tagok jelentése:
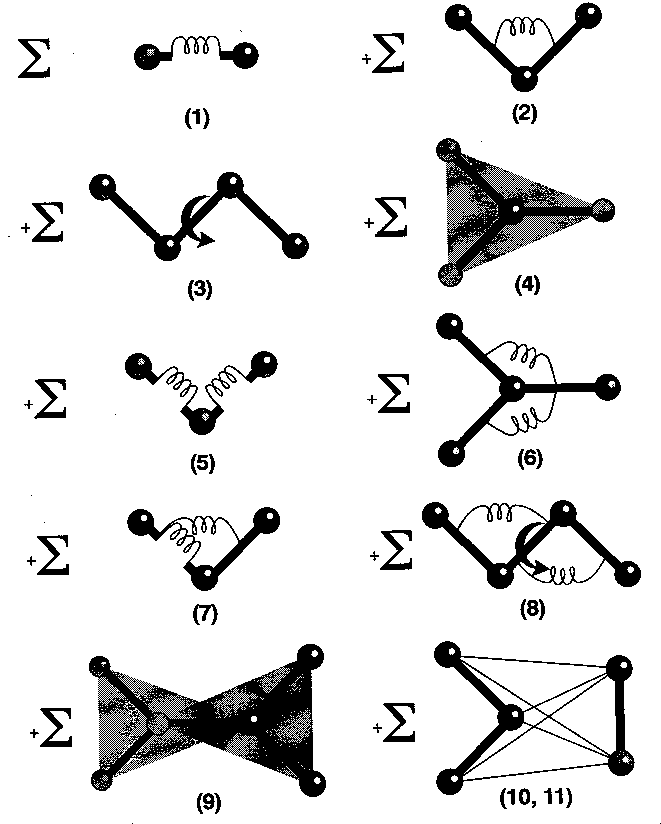
- 1. kötések nyújtása (Morse-potenciál vagy egyszerű harmonikus potenciál)
- 2. kötésszögek változtatása
- 3. torziós szögek változtatása
- out-of-plane kölcsönhatások (atom kitérése sík csoportból)
- 5-9. kereszttagok (csatolások az 1-4. effektusok között)
- 10. Van der Waals
- 11. Coulomb
- A képletben szereplő paraméterek értékét empirikus adatok (pl.
kristályszerkezet, rácsdinamika, röntgenadatok, sűrűség, párolgáshő, stb.)
alapján illesztik, néha ab initio kvantumkémiai számítások
eredményeit is felhasználják.
- Az empirikus adatok miatt a forcefield implicit módon magában foglalja a
relativisztikus és kvantummechanikai effektusokat is.
- Ismertebb forcefieldek fehérjékhez: AMBER, CHARMM, ECEPP, GROMOS, CVFF
A párkölcsönhatások problémája
- A párkölcsönhatások száma négyzetesen nő a rendszer méretével, ez nagyon
megnöveli az energiafüggvény kiértékeléséhez szükséges időt.
- Megoldás: cutoff (határtávolság) alkalmazása: a cutoffnál (10-20
angström) messzebb lévő párok kölcsönhatását nem vesszük figyelembe
(ez különféle problémákhoz vezet, amelyek azonban jól kezelhetőek).
Az oldószer modellezése
- Explicit: vízmolekulákkal körülvesszük a vizsgált molekulát (néhány
réteg vagy egy feltöltött doboz (periodikus határfeltétellel))
- Implicit: a potenciált módosítjuk, pl. távolságfüggő dielektromos
állandó, kétféle dielektromos állandó, stb.
Eljárások
Energiaminimalizálás
- Cél: a potenciálfelület minimumainak megkeresése (ezek körül fluktuál a
konformáció). Modellszerkezetek optimalizálására is jó.
- Legmeredekebb esés módszere: mindig az energiafelület deriváltjának
irányába lépünk.
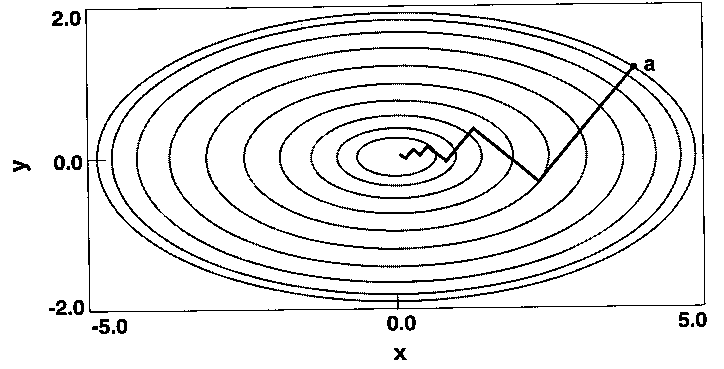
Az energiaminimumtól távol jól konvergál, hozzá közel rosszul.
- Konjugált gradiensek módszere: a lépésirányt az előző lépések
deriváltjainak felhasználásával korrigáljuk. Ez a módszer nagy rendszereknél
és a minimumok közelében is gyorsan konvergál.
Molekuladinamika
- Cél: a rendszer dinamikájának feltárása, konformációs mozgások szimulálása
- dt lépésköz: 1-2 femtoszekundum
- Módszer:
- Az atomoknak kezdeti sebességeket adunk (a kívánt hőmérsékletnek
megfelelő nagyságú és eloszlású atomi sebességek)
- A rendszer mozgását a Newton-féle mozgásegyenletek alapján szimuláljuk
(az energiafüggvényből számítjuk az erőket, ezekből a gyorsulásokat, a
sebességeket és a pozíciók változásait; többféle módszer)
- Végezhető szimuláció állandó hőmérsékleten (hőtartályhoz csatolt
rendszer) és állandó nyomáson
A fehérjemolekulák belső mozgásainak típusai
| A mozgás típusa | Térbeli kiterjedés
(angström) | Amplitúdó
(angström) | Jellemző időskála
|
|---|
| Kötések rezgései | 2-5 | 0,01-0,1 | 10-100 fs
|
| Globuláris régiók rugalmas deformációi | 10-20 | 0,05-0,5 | 1-10 ps
|
| Felszíni oldalláncok rotációja | 5-10 | 5-10 | 10-100 ps
|
| Eltemetett csoportok torziós rezgései | 5-10 | 0,5 | 10 ps - 1 ns
|
| Globuláris domének relatív mozgásai | 10-20 | 1-5 | 10 ps - 100 ns
|
| Belső oldalláncok rotációja | 5 | 5 | 100 mikrosec - 1 s
|
| Allosztérikus átmenetek | 5-40 | 1-5 | 10 mikrosec - 1 s
|
| Lokális legombolyodás | 5-10 | 5-10 | 10 mikrosec - 10 s
|
A molekuladinamika korlátjai
- A számítógépek jelenlegi teljesítőképessége mellett a szimulálható
időtartam korlátozott. Fehérjemolekulán az eddigi rekord 1 mikroszekundum
szimulációja (1 hónapig tartott egy 256 processzoros szuperszámítógépen).
Korszerű módszerekkel ez még növelhető kb. egy nagyságrenddel
- A klasszikus mechanikai modell nem alkalmas kémiai események
szimulálására (pl. ionizáció, protontranszfer, stb.)
Mire használható a molekuladinamika?
- Szerkezetfinomítás: még tökéletlen minőségű modellszerkezetek
finomíthatóak vele
- Szerkezetjóslás: mutáns fehérje szerkezete jósolható (kicseréljük
az oldalláncot, majd molekuladinamikával relaxáljuk a szerkezetet).
- Dokkolás: ligandum v. szubsztrát kötődési helyének megtalálása
egy fehérje felszínén (kevésbé megbízhatóan)
- Fehérjemolekulák fontos funkcionális mozgásainak feltérképezése:
pl. domének relatív mozgásai enzimkatalízis során, stb. (kevéssé megbízhatóan)
- Fehérjemolekulák le- és felgombolyodásának szimulációi: a
mechanizmus felderítése céljából
Molekuladinamikai programok
(Csak példák, sokkal több is van!)
Dokkolás

Benzamidin dokkolása tripszinhez
- Kismolekula (ligandum, szubsztrát, koenzim, stb.) kötődési helyének
megtalálása egy fehérje (receptor) felszínén
- Két fehérje egymáshoz való kötődési helyének megtalálása
Eljárás lényege: az egyik molekula mozgatása és forgatása a másik
felszínén, eközben az illeszkedés értékelése.
Az illeszkedés értékelésének módja szerint:
- Egyszerű geometriai illeszkedés figyelembe vétele
- Az illeszkedés kiértékelése bonyolult energiafüggvénnyel,
elektrosztatikus komplementaritás, stb.
A modell szerint:
- Mindkét molekula merev
- Az egyik molekula (ált. a ligandum) flexibilis, a másik (ált. a fehérje)
merev
- Mindkét molekula flexibilis (a keresés nagyon időigényes)
Az algoritmus szerint:
- Molekuladinamika
- Monte Carlo módszerek (pozíciók véletlenszerű generálása)
- Szimulált hőkezelés: szimuláció során magas hőmérsékletről indulva
lassan lehűtjük a rendszert, ez elősegíti az energiaminimum elérését
- Egyéb trükkök
Programok: DOCK,
AutoDOCK,
FTDOCK,
GRAMM, stb. (weben elérhetőek)
Eredményesség
- Kismolekula fehérjére dokkolásakor jó eredmények érhetőek el, de
bonyolultabb esetekben (pl. nagy fehérje, nagy és flexibilis szubsztrát)
csak kísérleti adatok ismeretében érhető el jó eredmény
- Fehérje-fehérje dokkolás: bizonytalan, gyenge eredmények
Bioinformatikai programcsomagok
A programcsomagok használatának előnyei a különféle webes
szolgáltatásokkal és különböző eredetű programokkal szemben:
- Egységes kezelés, ki- és bemenő formátumok
- Nem kell a szekvenciákat idegen szerverekre elküldeni (pl. ipari célú
kutatás esetén ez aggályos lehet).
- Kommerciális csomagok esetében van támogatás
Hátrányok viszont:
- Többnyire nincs meg néhány új, korszerű bioinformatikai eljárás
- Nagy hardver- és háttértárigény
Átfogó programcsomagok szekvenciaanalízisre
Wisconsin csomag (GCG)
- Kommerciális csomag, unixos nagygépekre
- Nem olcsó, de az EMBnet csomópontokon hozzáférhető (Magyarországon a
gödöllői Mezőgazdasági Biotechnológiai
Központnál igényelhető a hozzáférés).
- Parancssor üzemmódú programok, melyekhez van grafikus felhasználói
felület is (SeqLab)
- Több mint 130 program, a következő kategóriákban:
- Szekvenciák összehasonlítása:
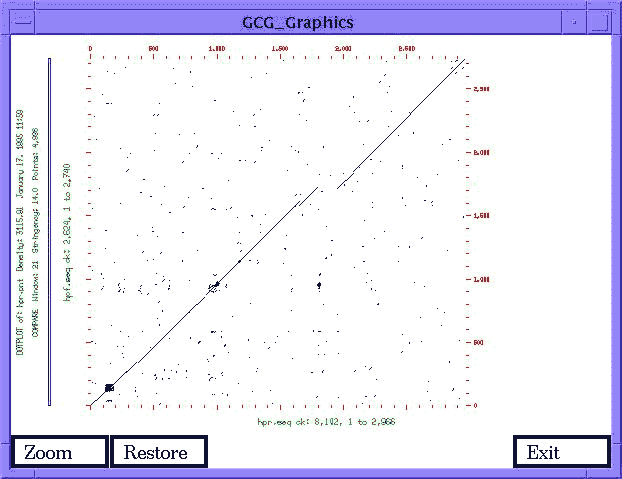
- Keresések adatbázisokban (BLAST, FASTA, Prosite-ban Motifs, stb.)
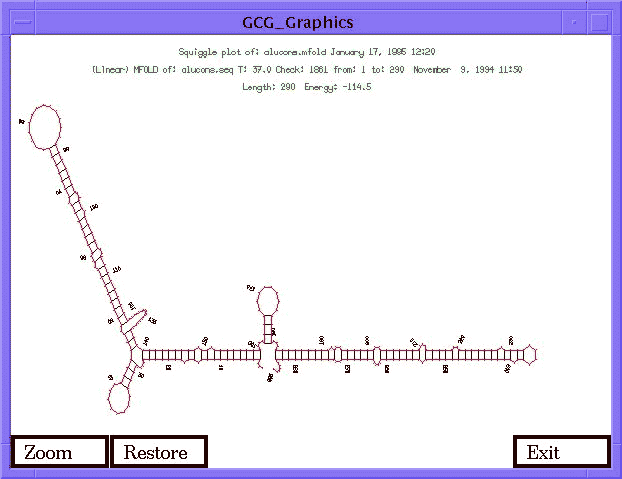
- RNS másodlagos szerkezetének jóslása:
- Szekvenciák szerkesztése, ábrakészítés. Plazmidtérkép:

- Filogenetikai analízis
- DNS-szekvenciák összeállítása fragmentumokból
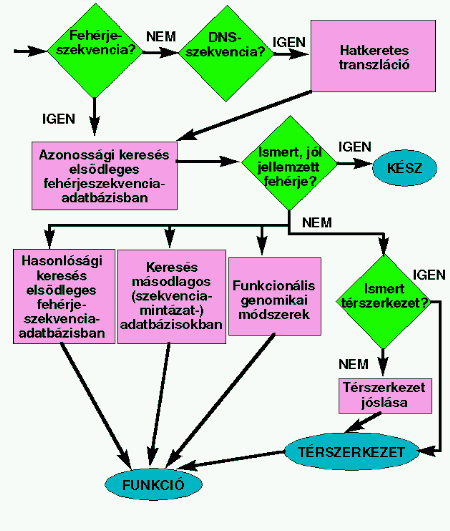
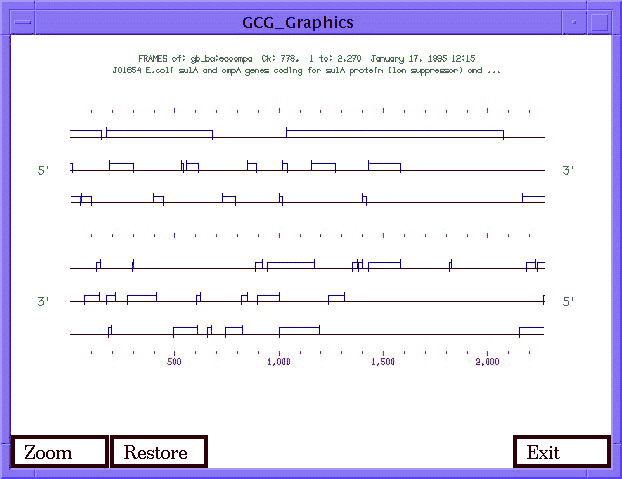
- Gének megkeresése, mintafelismerés. Hatkeretes transzláció:
- Különféle formátumú adatfájlok konverziója
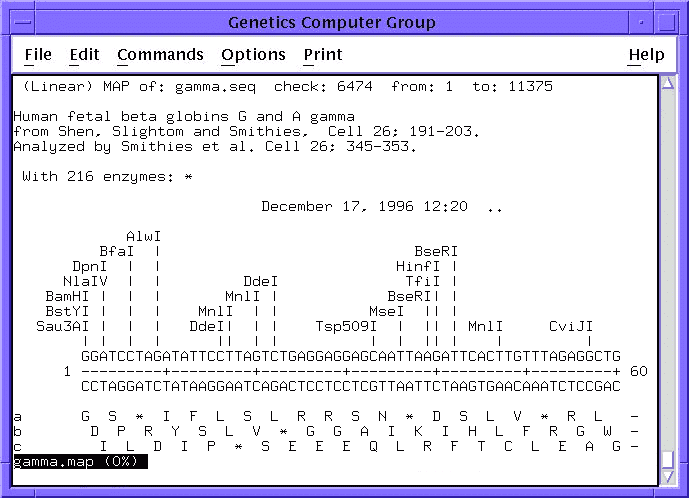
- Térképezés. Restrikciós enzimek hasítási helyei:
- Primertervezés (génklónozáshoz)
- Fehérjeszekvenciák elemzése. Pl. hélixkerék:
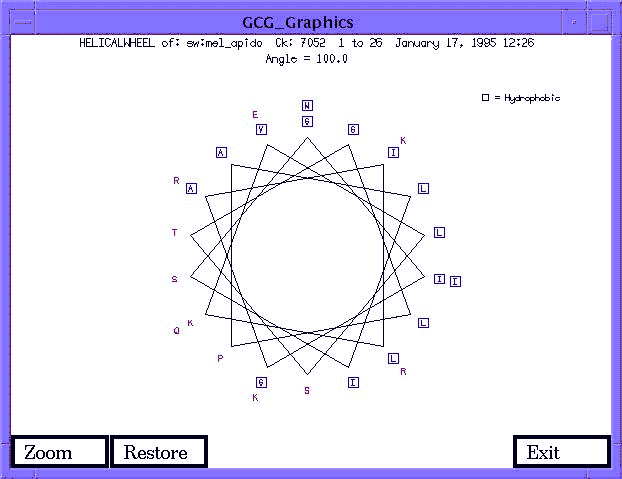
- Transzláció
- Akadémiai felhasználásra ingyenes, kommerciálisra pénzbe kerül
- UNIX/LINUX-ra. Windows változatok előkészületben.
- Sokféle program nukleotidszekvencia-analízisre
- Erőssége: a szekvenálást segítő programok
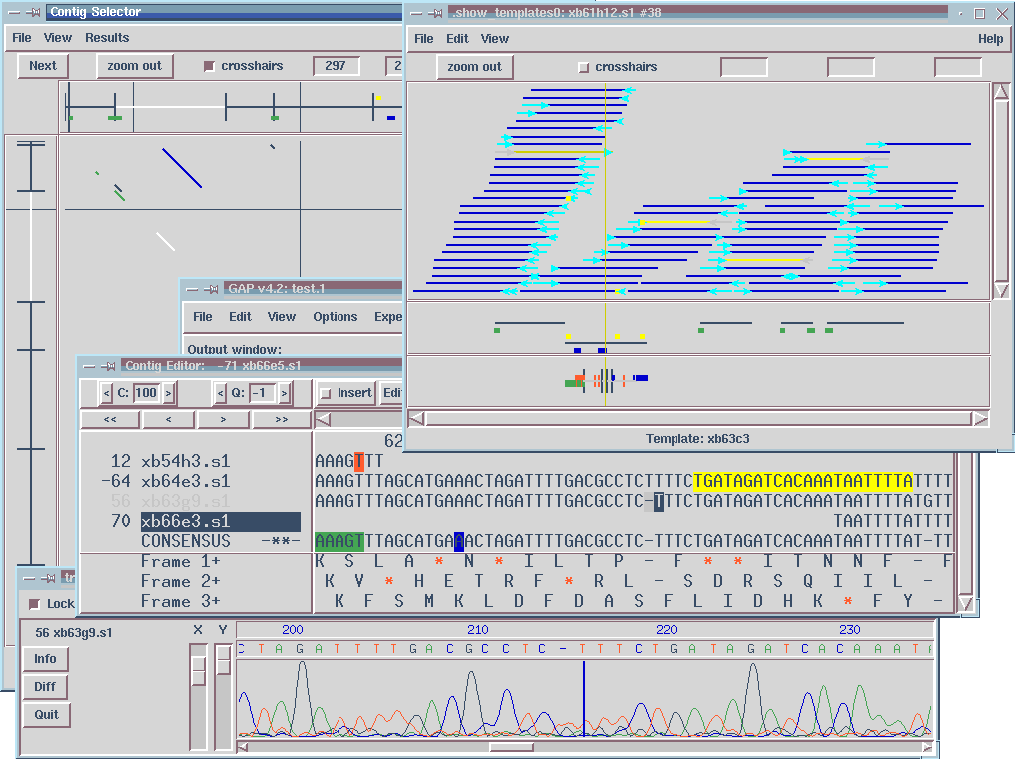
Szekvencia összeszerelése darabokból, a
fluoreszcens szekvenáló gél leolvasási adataival együtt
- Átfogó kommerciális csomag Windowsra és Macintoshra
- Moduljai:
- Szekvenciaösszeszerelés
- Gének megtalálása, annotáció
- Fehérjeszekvencia-elemzés
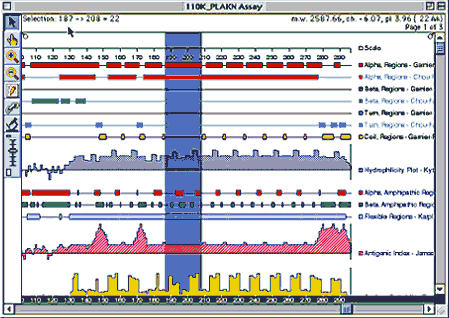
- Összerendezések
- Primertervezés
- Térképezés
- Szekvenciák szerkesztése
- Ingyenes, "Open source" programcsomag, folyamatos fejlesztés alatt
- A GCG-hez hasonló (annak bővítéseként keletkezett, majd függetlenné
vált), de grafikus felhasználó felület nélkül
- Kb. 100 program
Specializáltabb programcsomagok
- Összehasonlító genomanalízisre szolgál
- Ingyenes, fejlesztés alatt van, letölthető és Java verzió
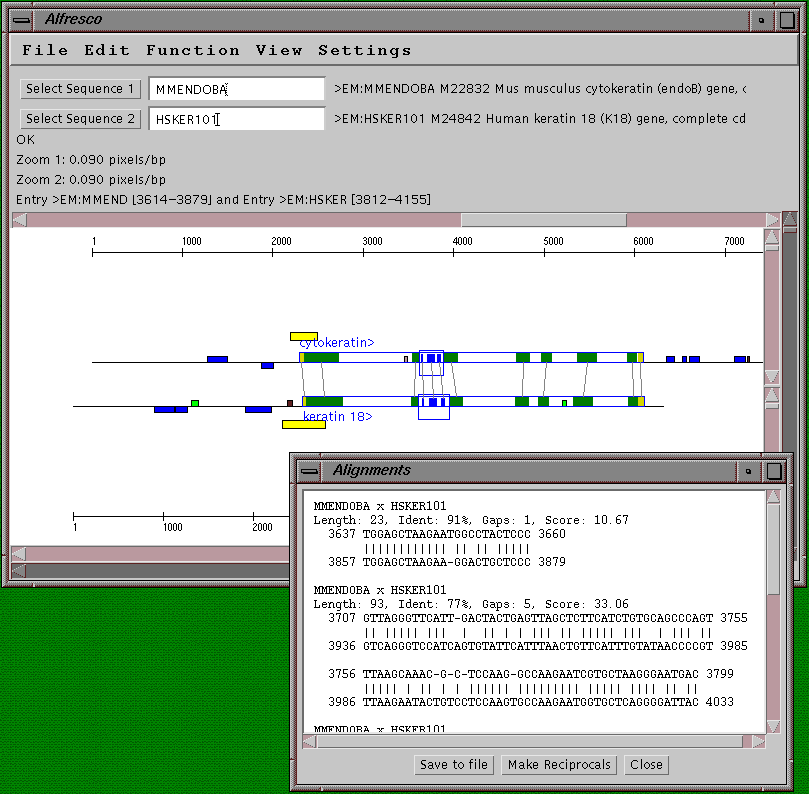
Az egér és az ember egy genomrészletének
összehasonlítása
- Elsősorban a molekuláris biológiai laboratóriumi munkákat könnyítik meg
(klónozás, primertervezés, PCR, stb.)
- Windows, Macintosh
"Bővített" szekvenciaeditorok
- A többszörös szekvencia-összerendezések szerkesztésére való programok
némelyike komolyabb szekvenciaelemző eljárásokkal is ki van egészítve, pl.
transzláció, dotplotok, hélixkerekek készítése, stb.
- Ingyenesek: BioEdit,
CINEMA, stb.
Bioinformatika a gyakorlatban
Az eddig tanultak szintézise.
A szekvenciaanalízis egy javasolt stratégiája