Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
10. Genomika 2.
- Microarray technikák és bioinformatikai vonatkozásaik
- Microarrayek és típusaik
- Korrelált génexpresszió mint a funkcionális genomika eszköze
- Kombinált megközelítés a funkcionális genomikában
- Genomikai adatbázisok
Microarray technikák és bioinformatikai vonatkozásaik
Microarrayek és típusaik
- Microarray (v. chip): kisméretű üveg- v. műanyag lap, melyre négyzetrács
szerinti elrendezésben biológiai mintákat visznek föl, minden pontba mást. A
vizsgált biológiai anyagot ezzel hozzák kölcsönhatásba, és valamilyen módon
detektálják, mely pontokban jött létre kölcsönhatás.
- Típusai:
- DNS microarray (oligonukleotid vagy cDNS)
- Peptid v. fehérje microarray
- Élő sejtek microarray-en (pl. élesztőtenyészetek)
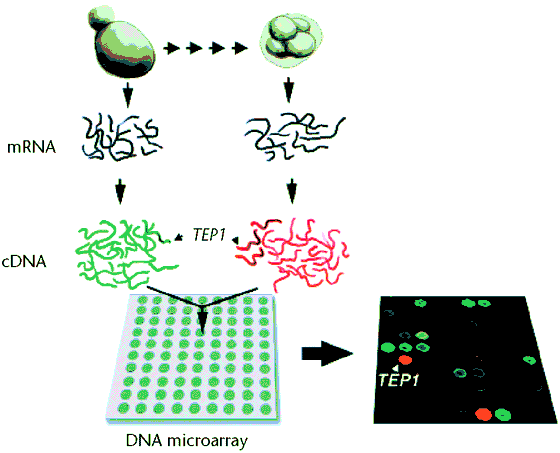
DNS microarray-ek
- Készítésük: az egyes DNS-darabokat robot helyezi el a megfelelő
helyekre, vagy helyben szintetizálják őket
- Nagy sűrűségűek: egy 1x1 cm méretű lemezkén több százezer pontban
helyezkedhetnek el a különböző DNS-ek
- Elhelyezhető rá pl. nagyszámú különböző gén cDNS-e, pl. az élesztő mind
a 6000 génje egyetlen lemezkén
- Alkalmazás: a fluoreszcensen jelölt vizsgált mintát (pl. cDNS-állomány)
hibridizáltatni próbáljuk a microarray-en lévő DNS-ekkel. A nem
hibridizáltakat lemossuk, fluoreszcencia detektorral leolvassuk, mely
pontokban történt hibridizáció
A DNS microarray-ek alkalmazásai
- A globális génexpresszió megfigyelése
- "Ujjlenyomat" készítése: az arrayre felvitt gének ismerete nélkül is
detektálhatjuk a génexpresszióban beálló változásokat
- A teljes genomot feldarabolva, genetikai különbségeket detektálhatunk
- stb.
Génexpresszió megfigyelése DNS microarray-ekkel
- Alapfeltevés: az mRNS-szint jellemzi az adott gén expressziós szintjét
és az adott fehérje mennyiségét is (nem teljesen igaz, de jó közelítés)
- A sejt különböző állapotaiban (pl. sejtciklus különböző fázisai, ill.
más-más környezet, tápanyagkészlet, stb.) vehetünk mRNS-mintát és microarray
segítségével jellemezhetjük az egyes gének expressziós szintjét.
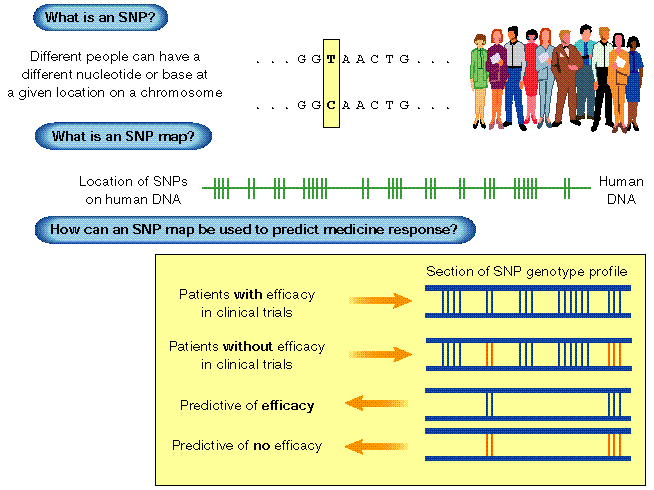
- Pl. két különböző állapot összehasonlító vizsgálata:
- Kétféle élesztősejt: vegetatív, ill. spóraképző állapotú
- mindkettőből kivonjuk a teljes mRNS-t, reverz transzkriptázzal cDNS-t
készítünk
- Az egyik mintát pirosan, a másik mintát zölden fluoreszkáló jelöléssel
látjuk el
- A kettő keverékét felvisszük a microarray-re, amely az összes
élesztőgén mintáját tartalmazza. A nem hibridizáltakat lemossuk
- Fluoreszcenciadetektorral leolvassuk, melyik pontra milyen mértékben
kötődött a pirossal, ill. a zölddel jelölt DNS.
- Ebből a kétféle állapotban expresszálódó gének azonosíthatóak
- A valóságos microarray képe (kinagyított részletekkel):

Korrelált génexpresszió mint a funkcionális genomika eszköze
- Az ugyanolyan körülmények között mindig együtt, azonos mintázat szerint
expresszálódó gének között funkcionális kapcsolat valószínűsíthető
- Bioinformatika újabb feladata: a microarray-adatok elemzése, kiértékelése
Génexpressziós profilok az élesztő sejtciklusának egyes fázisaiban
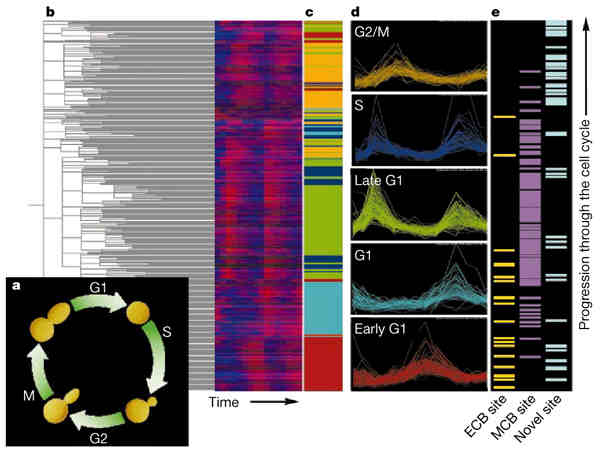
- Élesztősejteket szinkronizáltak (sejtciklusukat (a))
- Két ciklus során tízpercenként (18 alkalommal) vettek mintát a
sejtekből, melyek mRNS-állományából cDNS-t készítettek
- Ezeket az összes (6000) élesztőgént tartalmazó microarrayekkel
hibridizáltatták, így minden gén expressziós szintjét megmérték
- A 6000-ből 409 gén mutatott jelentős ingadozást az expressziós szintben,
ezeket vizsgálták tovább
- A 409 gént klaszterezték (csoportosították) az időbeli expressziós
mintázataik (ezek korrelációi) szerint (b: piros: nagy expresszió, kék: kis expresszió). A
fastruktúra (dendrogram) ezt a hierarchikus csoportosítást mutatja
- Időbeli expressziós viselkedésük (d) szerint a 409 gént 5 nagy csoportba
sorolták (c)
- (e: bizonyos gének helye a dendrogramban, most nem érdekes)
Az expressziós adatok klaszteranalízise
- Legfontosabb elemző eljárás
- Klaszterezés: egy halmaz elemeit egymáshoz való közelségük alapján
csoportokba soroljuk. Lehet egyszerű vagy hierarchikus:
Egyszerű klaszterezés
Hierarchikus klaszterezés
- Sokféle módszer, algoritmus
Egyéb elemző eljárások
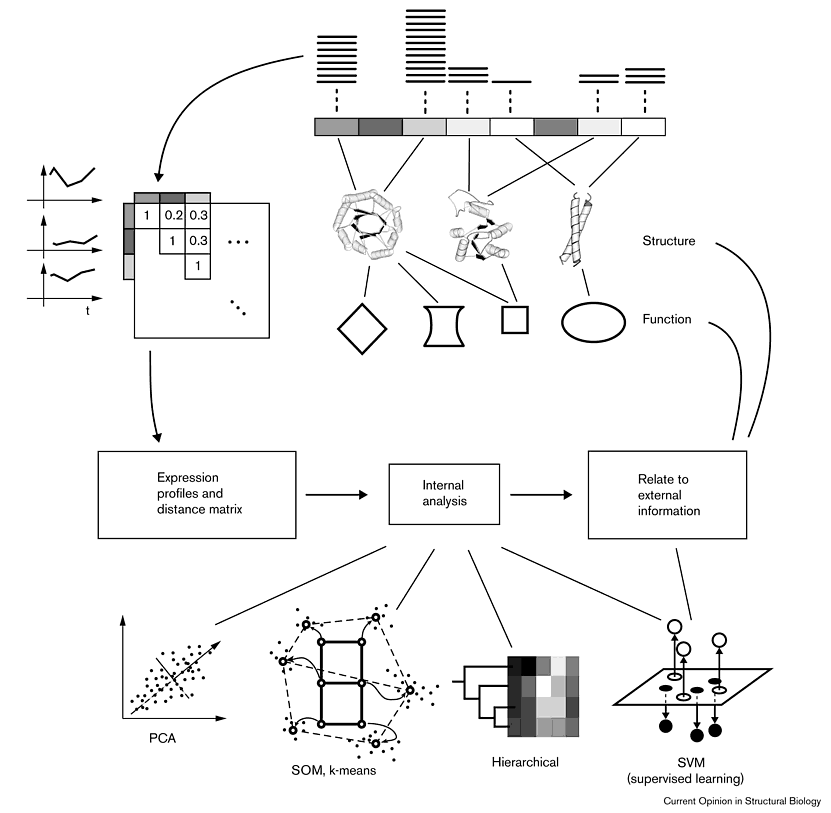
- Belső elemzés: csak magukat az expressziós adatok elemezzük,
máshonnan származó funkcionális v. szerkezeti információ felhasználása nélkül
- Főkomponens-analízis (principal component analysis, PCA)
- Egyszerű klaszterezési eljárások (SOM, k-means)
- Hierarchikus klaszterezés
- Külső elemzés: Az expressziós adatokat összefüggésbe hozza
funkcionális vagy szerkezeti információval
- SVM (support vector machine): matematikai eljárás, a megadott
funkcionális kategóriák szerint igyekszik szétválogatni az expressziós
adatokat
- Kiforratlan eljárások, sok nyitott kérdés (pl. nem világos, mi a jó
funkcionális csoportosítás)
Kombinált megközelítés a funkcionális genomikában
Az in silico funkcionális genomikai módszerek és a korrelált
génexpressziós adatok kombinálása a legeredményesebb.
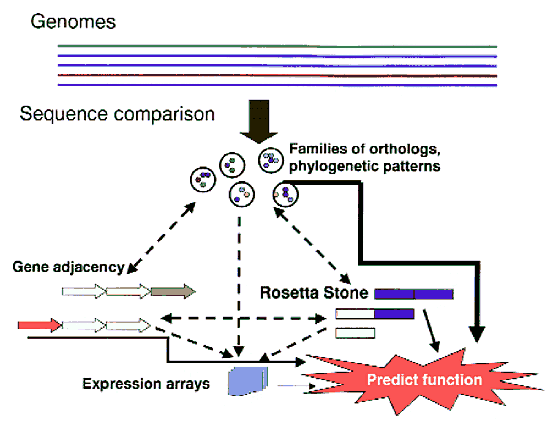
A nyilak vonalvastagságai a módszer megbízhatóságával arányosak
(legmegbízhatóbb a filogenetikai profilok módszere, legkevésbé a korrelált
expresszió)
Pl. az élesztőre elvégezve, a következő típusú eredmények adódtak:
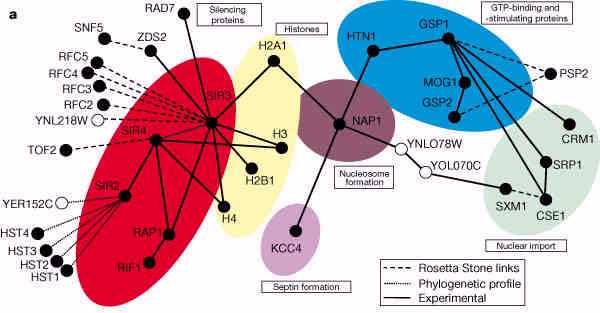
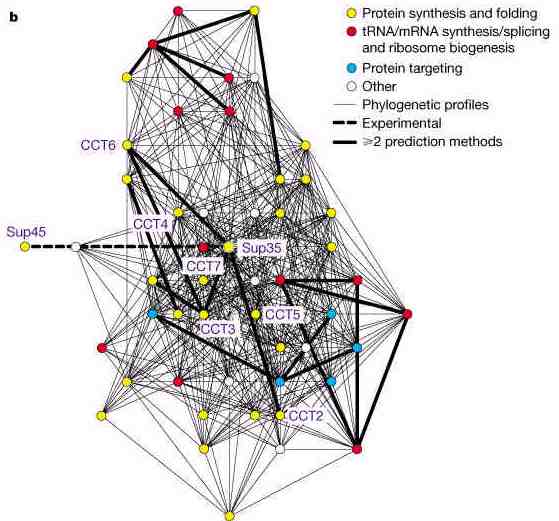
Sok a bizonytalanság, kísérleti megerősítés szükséges
Genomikai adatbázisok
Az új módszerek (teljes genomok szekvenálása, microarray módszerek,
proteomika) újfajta adatbázisok létrehozását teszik lehetővé.
DIP:
Database of Interacting Proteins (a kölcsönható fehérjék adatbázisa)
- Egymással kölcsönhatásba lépő (egymást kötő) fehérjékről gyűjt adatokat,
kísérleti eredmények alapján
- Kb. 6200 fehérjéről kb. 11 000 kölcsönhatást ír le
- Egy kölcsönhatás adatai: egyik fehérje, másik fehérje, kölcsönható
régiók, kísérleti módszerek, disszociációs állandó, irodalmi hivatkozások.
Példa.
- A kölcsönhatás-hálózatokat gráffal is szemlélteti (a csomópontok
kattinthatóak). Pl.:
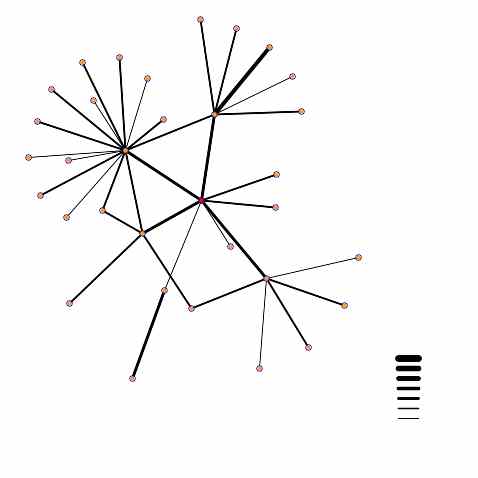
- Felhasználása: a meglévő adatok tárolása mellett új kölcsönhatások
megsejtésére is felhasználható
KEGG:
Kyoto Encyclopaedia of Genes and Genomes
Céljai:
- Számítógépesíteni a jelenlegi molekuláris és sejtbiológiai ismereteket,
adatbázisban tárolva az egymással kölcsönható, anyagcsere- vagy jelátviteli
útvonalhálózatokat és szereplőiket --> PATHWAY adatbázis
- Katalógusokat fenntartani az ismert teljes genommal rendelkező élőlények
génjeit, s a géntermékeket összekapcsolni a megfelelő
útvonalhálózat-komponenssel --> GENES adatbázis
- Adatbázisba foglalni az élő sejtekben előforduló összes kémiai
vegyületet, és ezeket összekapcsolni a megfelelő
útvonalhálózat-komponenssel --> LIGAND adatbázis
- Új bioinformatikai módszerek kidolgozása a funkcionális genomika
céljaira: útvonal-összehasonlítás, útvonal-rekonstrukció, útvonaltervezés
Séma:
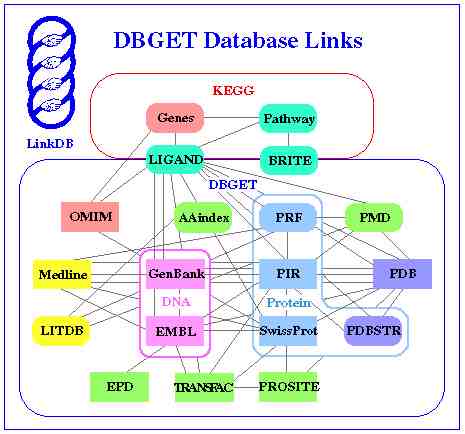
A KEGG a japán DBLINK (GENOMENET) adatbázisrendszerbe illeszkedik be.
Használat:
- útvonalak felől elindulva megkereshetjük az adott útvonalhoz tartozó
géneket
- gének felől elindulva az útvonalakat
- Műveletek útvonalakkal:
- Útvonal-rekonstrukció: az útvonalhálózat összeállítása az egyes
elemek közötti páronkénti kapcsolatokból. Ennek során fény derül az
esetlegesen hiányzó elemekre, ami új gének felfedezéséhez, ill. eddig
ismeretlen funkciójú gének funkciójának megjóslásához vezethet el.
- Útvonalak összehasonlítása: a különböző fajok
útvonal-hálózatainak egymásra illesztése alapján megtalálhatóak az
azonosságok és a különbségek, evolúciós események tárhatóak fel.
- Útvonalak elemzése: az útvonalak alapján számos új információ
nyerhető, pl. génduplikációk ismerhetőek fel, következtetni lehet a
génexpresszió szabályozására (pl. egy operonban elhelyezkedő géneknek az
útvonalhálózatban elfoglalt helye alapján, stb.)
- Új útvonalak tervezése: Összehasonlítások és elemzések alapján,
átlátva az egész útvonalhálózatot, lehetőség nyílik arra, hogy a hálózatot
valamilyen célnak megfelelően módosítsuk, pl. egy hatékonyabb növényvédőszer
vagy kevesebb mellékhatással bíró gyógyszer létrehozása érdekében.
Más, hasonló adatbázisok
- A KEGG-hez nagyon hasonló a WIT (What Is There) adatbázis
- Organizmusokra specifikus, részletesebb adatbázisok, pl. EcoCyc (E.
coli), stb.
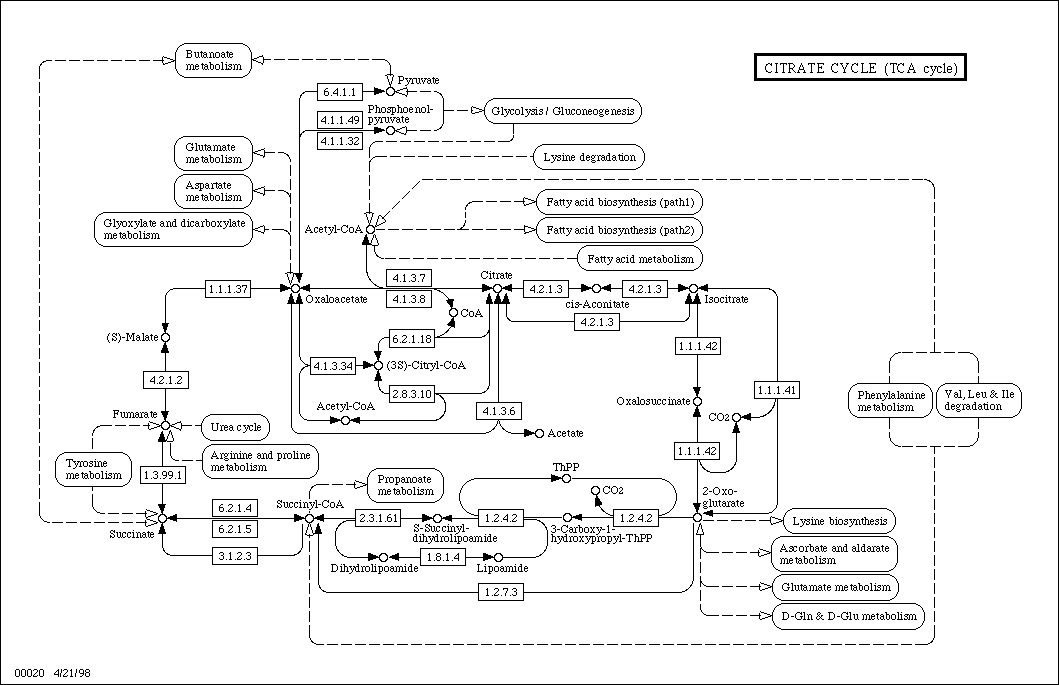
SNP: Single Nucleotide Polymorphisms (egynukleotidos polimorfizmusok
adatbázisai)
- A humán genomról van szó
- SNP: olyan pozíció a genomban, amelyen nemkonzerválódott nukleotid van.
Az 1%-nál ritkábban előforduló változatokat mutációknak nevezzük, ha
ennél gyakoribb változatok vannak, akkor polimorfizmusról beszélünk.
- SNP konzorcium: kutatók és cégek
társulása, az emberi genom SNP-inek felderítésére
- Emberi genomban becslés szerint 3 millió SNP hely van, ebből ma kb.
1 250 000 ismert (SNP adatbázisban).
Példa
Jelentősége
Linkage disequilibrium mapping
- Linkage disequilibrium (LD) ("a kapcsoltság egyensúlytalansága"):
azt mondjuk, hogy egy kromoszómán lévő két markerpozíció között linkage
disequilibrium áll fenn, ha a két pozíción található alléleket tekintve
bizonyos allélkombináció
gyakorisága eltér az egyes allélek gyakoriságának szorzatától.
Példa: két SNP pozíció, mindkettőn 50-50% gyakorisággal A, ill. G van, de a
kettőt nézve AG együtt fordul elő az esetek 40%-ában, 25% helyett. (Ha nincs
LD a két pozíció között, akkor az AG kombinációnak 50%x50%=25% gyakorisággal
kell előfordulnia.)
- Az LD oka: amikor az egyik helyen mutációként kialakul az egyik változat, a másik helyen
egy meghatározott változat van, így ez a kombináció rögződik.
- A generációk egymásutánjában a két pozíció közötti rekombinációk miatt a
kapcsolat az idő előrehaladtával egyre inkább fellazul, az allélkombináció
gyakorisága közeledik az egyensúlyi
értékhez (a két allélgyakoriság szorzatához)
- A két pozíció közötti rekombináció valószínűsége növekszik a pozíciók
távolságának növekedésével, ezért az LD mértéke a távolság növekedésével csökken
- Következmény: Betegséget, ill. gyógyszerre való
fogékonyságot/érzékenységet okozó (mutáns) gén közelében (ha a mutáció nem
nagyon régi eredetű) az SNP pozíciókban megfigyelhető allélgyakoriságok
eltérnek a normális embereknél megfigyelhető gyakoriságoktól, a mutáció és
az SNP hely közötti LD miatt!
- Az LD mértéke: valamely SNP allél gyakorisága a beteg/érzékeny/fogékony
emberekben mínusz ugyanez a normális emberekben.
- Mivel az LD mértéke nő a két pozíció közötti távolság csökkenésével, sok
SNP adatait felhasználva a betegséget/fogékonyságot/érzékenységet okozó gén
helye a kromoszómán behatárolható
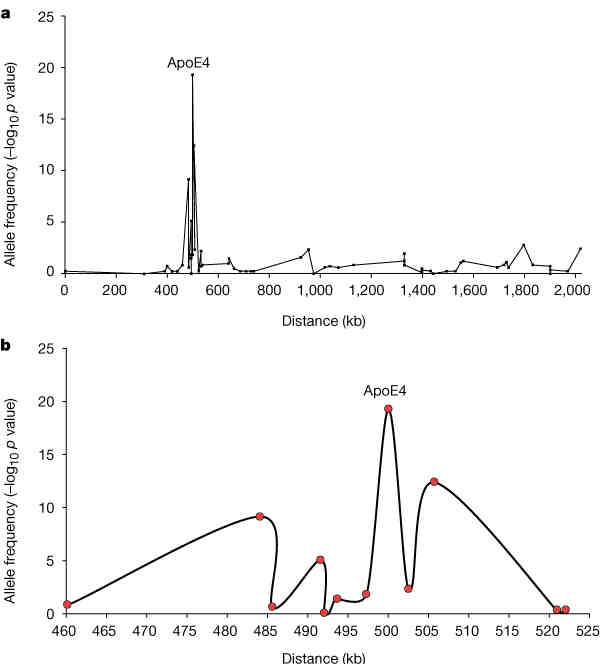
Az Alzheimer-kórért felelős egyik gén behatárolása LD
térképezéssel
- Minél sűrűbben helyezkednek el az SNP-k, annál jobb felbontással
határolhatjuk be a keresett géneket.