Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
8. A fehérjék térszerkezetének jóslása
A probléma bonyolultsága
- Általánosságban: találjuk meg egy tetszőleges szekvencia azon
konformációját, amely a szabadentalpia globális minimumát adja.
- Egyszerű modellekben kimutatható: a feladat ún. NP-nehéz, vagyis
a megoldásához szükséges idő a (fehérje)mérettel nempolinomiális függvény
szerint (hanem annál gyorsabban) növekszik. (Vagyis bizonyos mérethatár
fölött nem megoldható.)
- Gyakorlatban:
- a valódi fehérjék szekvenciái nagyon specifikusak
(evolúció során kiválogatódtak);
- a predikcióhoz felhasználhatjuk a már ismert térszerkezeteket mint
tudásbázist
- A gyakorlatban tehát a probléma kezelhető.
CASP-ok
- CASP: Critical Assessment of Methods of Protein Structure Prediction
(fehérjeszerkezet-jóslási módszerek kritikai felülvizsgálata):
kétévenként megrendezett "verseny" a predikcióval foglalkozó
kutatócsoportok között
- CASP1 (1994), CASP2 (1996), CASP3 (1998), CASP4 (2000).
- CAFASP1 (1998), CAFASP2 (2000): FA=Fully Automated: teljesen automatikus módszerek
ellenőrzése
- Forgatókönyv:
- Röntgenes és NMR-es csoportoktól begyűjtik azon fehérjék
szekvenciáit, melyek szerkezete várhatóan rövidesen ismertté válik
- a kutatócsoportok néhány hónap alatt ezekre predikciókat küldenek be
- decemberben (mikor már megvanak a kísérleti szerkezetek) összeülnek és
megbeszélik az eredményt; a legsikeresebb
jósok előadást tartanak; a Proteins c. folyóirat különszámot ad ki (ált. a
következő év októbere táján, a CASP4 eredményeit még nem közölték).
Három fő módszer
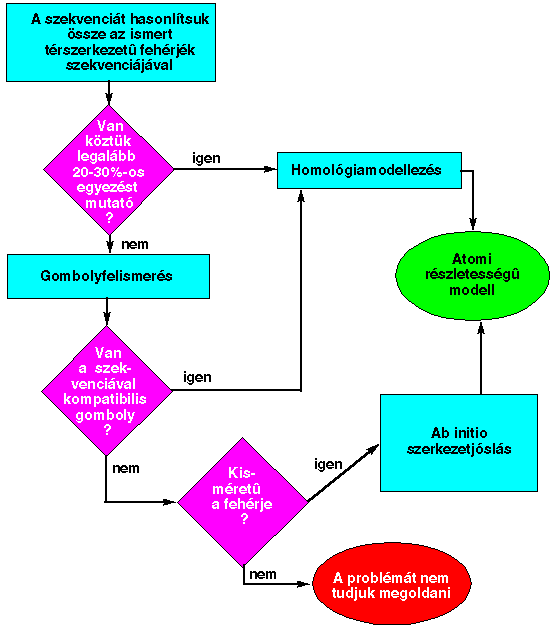
- Homológiamodellezés (komparatív modellezés): ha a
szekvenciánkhoz van
ismert térszerkezetű, a szekvenciánkkal számottevő szekvenciaazonosságot
(> 20%) mutató homológ, akkor annak térszerkezete alapján építhetjük fel
a mi szekvenciánk jósolt térszerkezetét.
- Gombolyfelismerés (fold recognition): alacsony
szekvenciaazonosság mellett meg kell találni, van-e az ismert
térszerkezetek között a szekvenciánkkal kompatibilis gomboly, s melyik az.
Ha találtunk ilyet, akkor homológiamodellezéssel szerkezet is építhető.
- Ab initio predikció: nincs számottevő szekvenciaazonosság
ismert térszerkezetű fehérjével, s nincs kompatibilis gomboly sem, tehát a
szekvenciánkhoz tartozó gomboly ismeretlen. A térszerkezet-predikció
ilyenkor fizikai elvek felhasználásával történhet.
Emlékeztető: A fold ("gomboly") fogalma
- Fold: Egy fehérje nagybani, durva szerkezete, a polipeptidlánc
gerincének durván vett térbeli lefutása. Magában foglalja a másodlagos
szerkezeti elemek körülbelüli, relatív elhelyezkedését és
összeköttetéseik sorrendjét. A folding (felgombolyodás)
szóból. Magyarul gombolynak mondhatnánk.
- A hasonló szerkezetű fehérjéknek ugyanaz a foldja, vagyis a gombolya. A
fold tehát egy fehérjecsaládot határoz meg, szerkezeti hasonlóság alapján.
Példa:
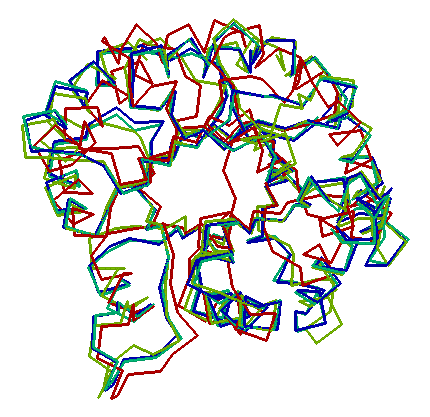
| 
|
| Különböző fajokból származó trióz-foszfát izomerázok
(TIM) és hasonló fehérjék egymásra illesztett szerkezete (alfa-szénatomokból
álló váz).
| A fehérjecsalád fold-ja, azaz "gombolya", az ún.
TIM-barrel fold
|
A predikció várható sikeressége
- A szekvencia-adatbázis gyorsabban növekszik, mint a térszerkezeti
adatbázis
- Térszerkezet jobban konzerválódott, mint a szekvencia: két fehérje
30%-os szekvenciaazonossága esetén térszerkezetük nagyon hasonló. 20-30%
között (szürkületi zóna) a hasonlóság kérdésessé válik, de még 10%
alatti szekvenciaazonosság esetén is előfordulhat
- Térszerkezet-jóslás legjobban használható módszerei:
- Homológiamodellezés: ha az ismeretlen térszerkezetű fehérje
szekvenciája >20-25% azonosságot mutat egy már ismert térszerkezetűével,
akkor jó közelítő modell építhető
- "Gombolyfelismerő" (fold recognition) eljárások: az esetek egy
részében kisebb szekvenciaazonosság esetén is azonosítják a "gombolyt",
ezután pedig a homológiamodellezés már alkalmazható a megtalált hasonló
térszerkezet felhasználásával.
- Adott szekvencia mekkora valószínűséggel modellezhető a fenti
eljárásokkal? Attól függ, honnan vesszük a szekvenciát:
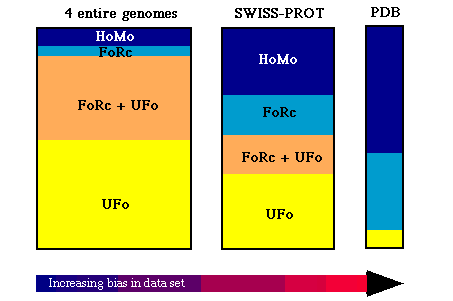
(HoMo: homológiamodellezés, FoRc: fold recognition [gombolyfelismerés],
UFo: unknown fold [ismeretlen gomboly], UFo+FoRC: határeset)
- PDB erősen redundáns: a benne lévő szerkezetek 58%-a modellezhető
lenne más ismert szerkezetek alapján
- SWISS-PROT szekvenciaadatbázis szintén torz: a benne lévő
szekvenciák kb. 30%-ára lehetne homológiamodellezéssel modellt építeni
- Újonnan megszekvenált genomok fehérjéi: csak kb. 10%-ra lenne
építhető homológiamodell; kb. 40% felismerhető lenne
gombolyfelismeréssel, a többi 50% teljesen új szerkezet
- Szerkezeti genomika: célja, hogy a genomban kódolt fehérjék közül
kiválogassa azokat a fehérjéket,
amelyek szerkezetét kísérletesen (röntgen, NMR) meghatározva az összes
többi fehérje homológiamodellezhetővé válik
Homológiamodellezés
A homológiamodellek rendkívül hasznosak kísérletek tervezéséhez,
hipotézisek felállításához, stb.
Hagyományos módszer: lánctöredékek összeszerelése
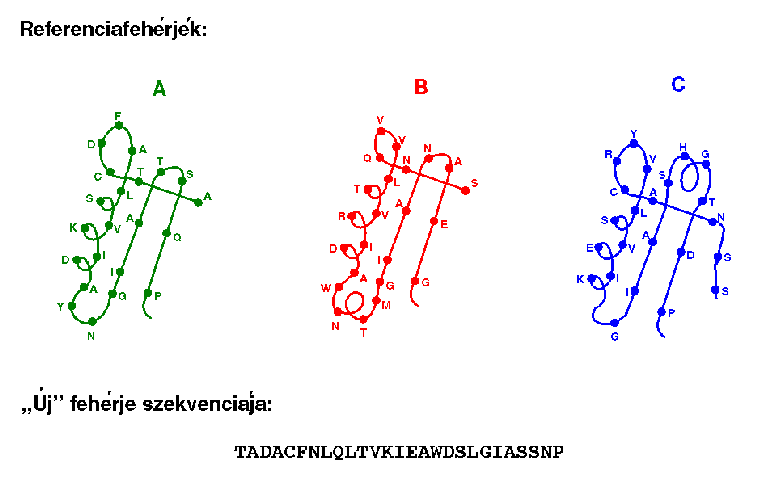
Kiindulás:
- referenciafehérjék (más néven: templátok vagy
anyaszerkezetek): a modellezendő fehérje szekvenciájával jelentős
azonosságot mutató fehérjék ismert szerkezete
- "Új" fehérje (más néven: célfehérje): a modellezendő fehérje,
melynek csak a szekvenciája ismert
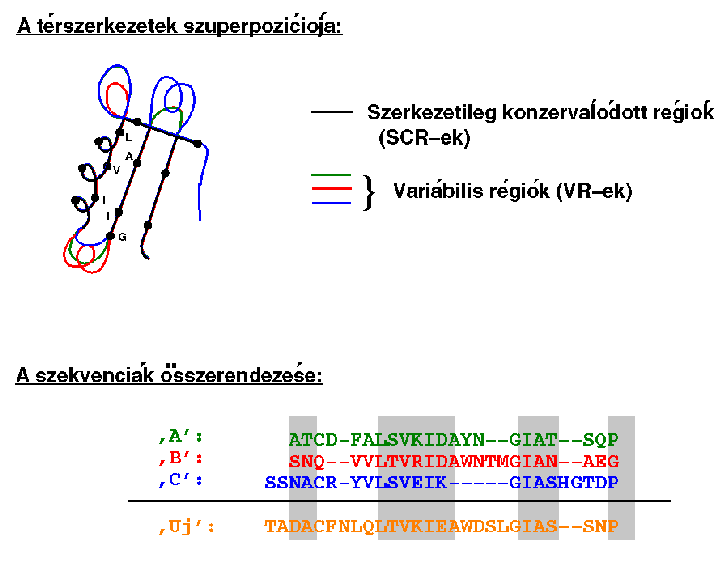
A három fehérjét egymásra illesztjük, így kitűnik, melyek a
szerkezetileg konzerválódott régiók (SCR-ek) és a variábilis
régiók (VR-ek). A három fehérje szekvenciáját a térszerkezetek fedése
alapján összerendezzük, majd hozzárendezzük az új fehérje szekvenciáját:
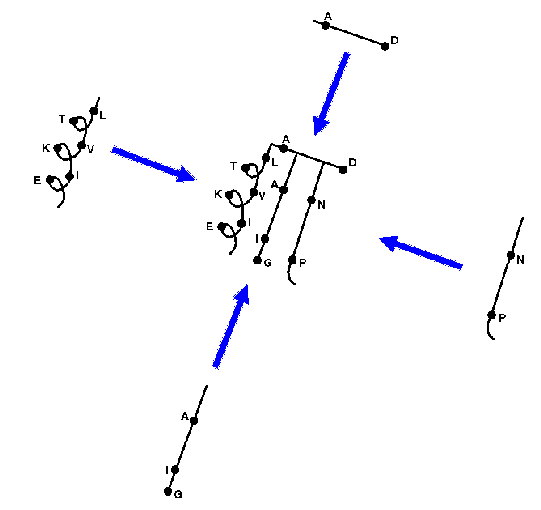
Az új fehérje felépítése: az SCR-eket bármelyik referenciafehérjéből
átvehetjük:
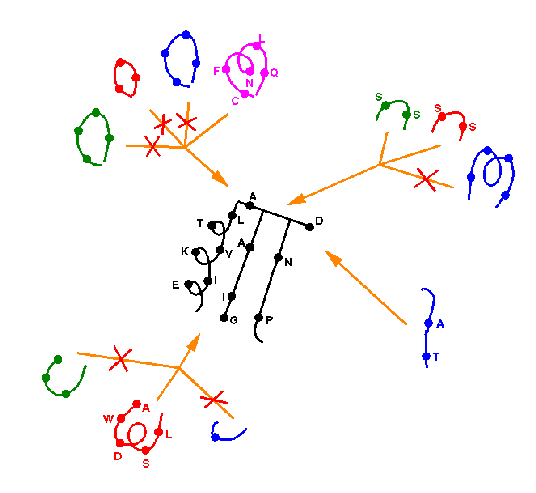
A VR-eket egyenként megvizsgáljuk. A referenciafehérjék megfelelő
VR-jei közül kiválasztjuk a legilleszkedőbbeket. Ha nincs megfelelő,
akkor adatbázisban keresünk odaillő hurkot.
Így előáll a durva modell, mely darabokból van összeszerelve.
Energiaminimalizálással finomítható.
- Oldalláncok modellezése: az oldalláncokat ki kell cserélni a
célfehérjének megfelelőre. Konformációjuk beállítása: a
templátfehérjékből átvenni, amennyire lehet, ill. különféle
optimalizálási eljárások (pl. rotamerkonformációkkal)
- Hurkok modellezése: ha a templátfehérjék között nincs megfelelő,
akkor hurokadatbázisban keresünk megfelelőt, vagy valamilyen
konformációkereső eljárást alkalmazunk
- A modell pontossága: az SCR-ek közepén a legpontosabb, a
hurkokban a legpontatlanabb
A térbeli kényszerek kielégítésén alapuló módszer
- MODELLER program (Andrej Sali)
- A templátszerkezetekből térbeli kényszereket vezet le a
célszerkezetre vonatkozóan (pl. atompárok távolsága, atomhármasok szöge,
stb.)
- A kényszereket egy célfüggvénybe egyesíti, majd random kiinduló
szerkezetből kiindulva szimulált hőkezeléssel optimalizálja
- Jó minőségű szerkezeteket állít elő, kevés sztereokémiai hiba
Homológiamodellezés a CASP3-on
- Alkalmazott módszerek: a fenti módszerek kombinációi, speciális
algoritmusokkal bővítve, sok helyen emberi beavatkozással
- Sikeresség:
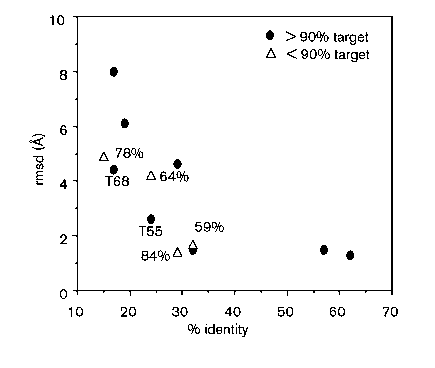
X: szekvenciaazonosság a templáttal, Y: rmsd a valódi szerkezettől
rmsd: root mean square deviation, két szerkezet
egymástól való különbözőségének mértéke, tkp. az egymásnak megfelelő atomok
közötti távolságok négyzetes közepe.
Látható: kb. 30% szekvenciaazonosság fölött igen jó modell építhető
- A nagyobb, templát nélküli hurkok és az oldalláncok modellezése nem
kielégítő pontosságú
- Legkritikusabb tényező: a szekvencia-szerkezet összerendezés.
- Az összerendezést nagyon gondosan kell elkészíteni, pl. figyelembe
véve a templátszerkezetekben konzerválódott H-kötéseket, kontaktusokat,
stb.
- Az elkészült modellt megvizsgálva az összerendezésen javítani
lehet, s annak alapján jobb modellt építeni
Gombolyfelismerés
- Távoli homológok (<25% szekvenciaazonosság) gyakran azonos
gombollyal rendelkeznek. A feladat ennek felismerése
- Két dolog kell hozzá:
- Egy "gombolykönyvtár", ami az ismert térszerkezeteket (gombolyokat)
tartalmazza valamilyen formában.
- Egy összehasonlító módszer, amellyel meg tudjuk állapítani, hogy egy
adott szekvencia mennyire illeszthető, húzható rá egy adott térszerkezetre
- A módszer: a szekvenciánkat egyenként az összes gombollyal
összehasonlítjuk, hogy megtaláljuk, van-e köztük olyan, amit a szekvenciánk
felvehet.
- Általános eljárás: felfűzés (threading): az ismeretlen
szerkezetű fehérje szekvenciáját valamiképpen "fel kell fűzni" az
ismert térszerkezetekre, és valamilyen potenciálfüggvénnyel értékelni
kell a szekvencia és a szerkezet illeszkedését
- Korábbi eljárások: az oldalláncok környezetét vették alapul
(környezet polaritása, eltemetettség, másodlagos szerkezet, stb.)
- Újabb eljárások: a párpotenciálokon van a hangsúly (az ismert
térszerkezetekben található aminosav-aminosav kontaktusok alapján
levezetett potenciálfüggvények)
Felfűzés a CASP3-on
- A hat legeredményesebb csoport módszerei:
- Három csoport (Jones, Sippl, Bryant) párpotenciálokon alapuló pontozófüggvényt
alkalmazott, kiegészítésekkel:
- Jones csoportja és Sippl csoportja (ProFIT program): a potenciálfüggvény az
aminosavpárok szekvenciabeli és térbeli távolságától is függ; továbbá a
jósolt másodlagos szerkezetet is figyelembe veszik
- Bryant: a kontaktpotenciálokon felül az ismert térszerkezetekben
talált, konzerválódott hidrofób magok ismeretére is támaszkodott
- K. Karplus: tisztán szekvencia alapú módszer, ún. rejtett Markov-modell
- Nishikawa, Koretke csoportjai: sokféle módszer kombinációját
alkalmazták, ezek alapján konszenzus
- Eredmények:
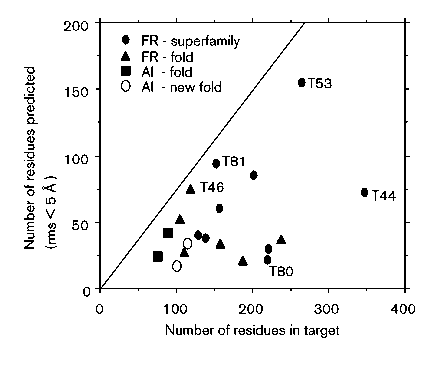
A legjobb gombolyfelismerési predikciók. X: a fehérje hossza; Y: az
5 angströmnél kisebb rmsd-vel (a valódi szerkezettől való eltérés)
prediktált aminosavak száma
- Látható: az eredmény vegyes. Néhány fehérjére jó a predikció, a
többségre elég rossz, némelyikre csapnivaló
- A felfűzési módszerek egyelőre nem kielégítőek (egyetlen módszer sem
képes az esetek >40%-ában helyes eredményt adni)
Távoli homológiák felismerése
Ha szekvenciahasonlóság alapján a szekvenciánkhoz nem találunk ismert
térszerkezetű homológot, akkor segíthet egy olyan módszer, amely távoli
homológokat is megtalál, s ezek között lehet egy olyan, ismert térszerkezetű
homológ, amelyet
fel tudunk használni a térszerkezet-jósláshoz. A gombolyfelismerés is tkp.
távoli homológiát detektál (felismeri a rokonságot két fehérje között az
alacsony szekvenciabeli hasonlóság ellenére).
- A probléma másik megközelítése: tisztán a szekvenciák alapján dolgozunk
- PSI-BLAST program (Position Specific Iterated Basic Local
Alignment Search Tool): a BLAST összerendezés-kereső program
kiterjesztése:
- Előbb egy hagyományos BLAST kereséssel kigyűjti egy szekvencia
homológjait, ezekből többszörös összerendezést készít
- A többszörös összerendezésből elkészít egy szekvenciaprofilt
- A szekvenciaprofillal újból keresést végez a szekvenciaadatbázison
- Ezáltal távolabbi homológokat is megtalál
- A PSI-BLAST a felfűzési módszerekkel összemérhető hatásfokkal
találja meg a távoli homológokat, így a gombolyfelismerés versenytársa lehet.
Ab initio térszerkezetjóslás
CASP3
A célszerkezetek:
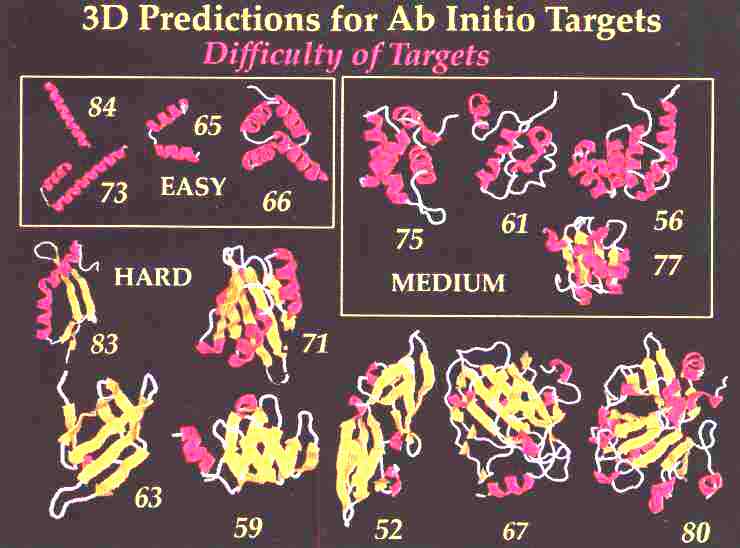
Az alkalmazott módszerek
Nincs standard eljárás. Sokféle, egyedi módszer:
- Skolnick és mtsai: többszörös szekvenciaösszerendezésekből
kényszerfeltételeket vezettek le a
másodlagos és harmadlagos szerkezetre [kontaktusok] nézve. Ezután rácsmodellként
szimulálták a fehérje felgombolyodását; 1000 szimuláció közül a
legalacsonyabb energiát eredményezőt tekintették végleges modellnek
- Scheraga és mtsai: "egyesített-atom" modellben (csak
alfa-szénatomok) konformációkeresés, majd "összes-atom" modellben
finomítás
- Osguthorpe: egyszerűsített fehérjemodell, molekuladinamikai
szimulált hőkezelés
- Levitt és mtsai: tetraéderes rácsmodellként generálták az
összes lehetséges konformációt, majd a legalacsonyabb energiájúakat alapul
véve, másodlagosszerkezet-jóslást is felhasználva, áttértek
"összes-atom" modellekre, ezeket egy komplex pontozófüggvénnyel értékelték
- Mosberg és mtsai: másodlagosszerkezet-jóslás után manuálisan
illesztgették össze a másodlagos szerkezet elemeit, hogy eltemetődjenek
a hidrofób felszínek
- Baker és mtsai: 3-9 aminosav hosszúságú fragmentumokra
kerestek modelleket az ismert szerkezetek adatbázisában, majd ezeket
összeillesztették, egy energiafüggvénnyel értékelték, majd a szemre
legjobban kinéző mellett döntöttek
Eredmények
- Több csoportnak sikerült >30 aminosavas fragmentumokat jó
közelítéssel jósolni (< 4 angström rmsd)
- Az alfa és az alfa-béta típusú fehérjék architektúráját többnyire
jól megközelítették
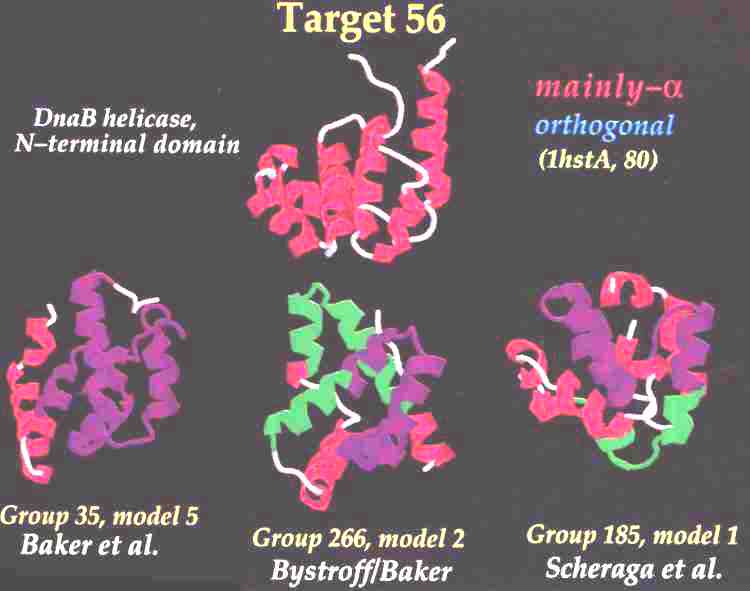
- A tudás alapú (ismert szerkezetek adatbázisára támaszkodó) módszerek
jobban teljesítettek, mint a fizikai alapú módszerek
- Egészében véve komoly előrelépés van a CASP2-höz képest
CAFASP1
- Teljesen automatikus gombolyfelismerési módszerek tesztje
- Eredmény: jóval gyengébben működnek, mint emberi beavatkozással
További kilátások
- CASP4: már lezajlott, de még nem közölték az eredményeket.
- Nagyobb szabású predikciós projektek kezdődtek a szerkezeti
genomikai programok kapcsán
- Már van MODBASE modelladatbázis, mely folyamatosan bővül. A
kísérletes szerkezetek meghatározásával párhuzamosan ellenőrizhetők a
modellek