Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
7. Fehérjeszekvenciák és -térszerkezetek analízise.
1. Egyszerű elemzések
2. Térszerkezet-predikció
2.1. A probléma bonyolultsága
2.2. A predikció szintjei
2.3. 1D predikciók (másodlagos szerkezet, hozzáférhetőség, transzmembrán
hélixek
3. Térszerkezetek elemzése:
3.1. Minőségellenőrzés
3.2. Másodlagos szerkezet
3.3. Szerkezeti motívumok
3.4. Kölcsönhatás ligandumokkal
3.5. Töltésviszonyok
3.6. Felszínek, üregek
Egyszerű elemzések (lásd Expasy Tools)
Fehérjeazonosítás
- AACompIdent: fehérje azonosítása aminosav-összetétel alapján.
Megadható még az izoelektromos pont (pI) és a molekulatömeg (Mw). Eredmény:
rangsorolt lista a SWISSPROT-ban lévő, a megadott adatokhoz közeli
paraméterekkel rendelkező fehérjékről. Távoli homológiák detektálhatók
ezen a módon!
- PROPSEARCH: A megadott szekvenciából (csak az
aminosav-összetételből) 144 tulajdonságot számít ki (pl. nagy és kis
oldalláncok részaránya, átlagos hidrofobicitás, átlagos töltés, bizonyos
dipeptidek gyakorisága, stb.) Ezek alapján keres hasonlót a
szekvenciaadatbázisokban. Távoli homológiák detektálhatóak vele!
Fehérjeazonosítás tömegspektrometriai eredmények alapján
Tömegspektrometria: valamilyen specifikus proteázzal (pl. tripszin) való
emésztés után megadja a kapott peptidek pontos molekulatömegét. Ennek
alapján a fehérje azonosítható.
Leggyakrabban használt módszer: MALDI (Matrix Assisted Laser
Desorption/Ionisation Mass Spectrometry)
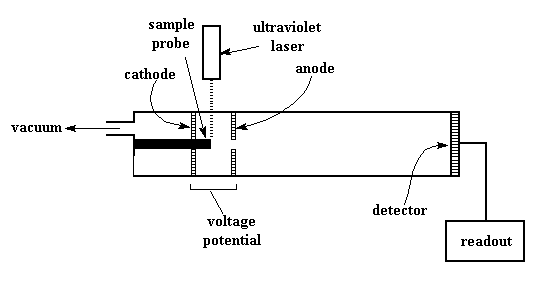
A fehérjét (annak fragmentumait) UV-elnyelő anyagba ágyazzák (mátrix), ezt a réteget UV
lézerrel lövik, mire a molekulák ionizálódnak és leválnak a mátrixról.
Elektromos térben felgyorsulnak, detektor méri a becsapódást. A becsapódásig
eltelt időből számítható a tömeg. Spektrum:
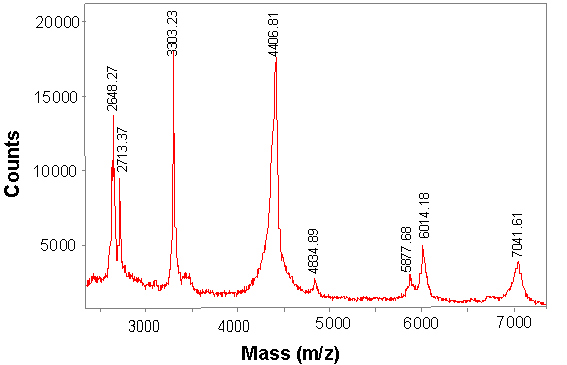
- MOWSE: megadva az emésztéshez használt proteázt és a
peptidtömegeket, rangsorolt listát ad az OWL adatbázisban lévő, a megadott
adatoknak megfelelő fehérjékről
Fizikai tulajdonságok a szekvenciából
- ProtParam: a szekvenciából egy sor mennyiséget számít ki (Mw, pI,
aminosav- és atomösszetétel, extinkciós koefficiens, stb.)
- PeptideMass: a megadott szekvencia proteolitikus emésztési
fragmentumainak tömegét számítja ki
- SAPS (Statistical Analysis of Protein Sequences): rengeteg
fizikai és kémiai információt számít a szekvenciából (aminosav-összetétel,
töltéseloszlás, pozitív és negatív töltésű klaszterek, mintázatok, hidrofób
szegmensek, repetitív régiók, periodicitás, stb.)
Térszerkezet-predikció
A probléma bonyolultsága
- Általánosságban: találjuk meg egy tetszőleges szekvencia azon
konformációját, amely a szabadentalpia globális minimumát adja.
- Egyszerű modellekben kimutatható: a feladat ún. NP-nehéz, vagyis
a megoldásához szükséges idő a (fehérje)mérettel nempolinomiális függvény
szerint (hanem annál gyorsabban) növekszik. (Vagyis bizonyos mérethatár
fölött nem megoldható.)
- Gyakorlatban:
- a valódi fehérjék szekvenciái nagyon specifikusak
(evolúció során kiválogatódtak);
- a predikcióhoz felhasználhatjuk a már ismert térszerkezeteket mint
tudásbázist
- A gyakorlatban tehát a probléma kezelhető.
A predikció szintjei
Jósolhatóak a fehérjeszerkezet egy-, két-, ill. háromdimenziós aspektusai:
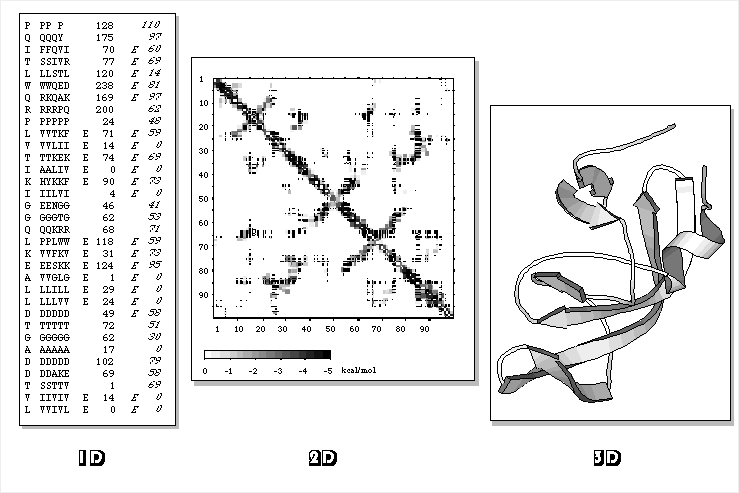
(1D, 2D és 3D információ. A 2D a kontaktustérkép.)
- 1D: aminosavakhoz köthető tulajdonságok, melyek 1D stringként
írhatóak fel. Pl.: szekvencia, másodlagos szerkezet, oldószer általi
hozzáférhetőség, hidrofobicitás
- 2D: aminosavpárok közötti távolságok, kontaktusok
- 3D: az összes atomi koordináta
1D predikciók
Másodlagos szerkezet jóslása a szekvenciából
A fontosabb módszerek az Expasy -
Tools oldalról elérhetőek.
Módszerek (rengeteg van, ez csak néhány példa):
| Módszer | Működés elve | Kb. pontosság (%)
|
|---|
| 1. generációs | Chou-Fasman (CF) | Az egyes
aminosavtípusok előfordulásának valószínűsége a különböző másodlagos
szerkezeti elemekben | 55
|
| Garnier-Osguthorpe-Robson (GOR I, GOR II) | 56
|
| Lim | 57
|
| 2. generációs | Nagano | Statisztikai
adatok (aminosavpárok, ill. triplettek) | 61
|
| GOR III | 62
|
| Ptitsyn-Finkelstein | Fizikai-kémiai tul. | 61
|
| Qian-Sejnowski | Neuronhálózat | 60
|
| GOR IV | Szegmensstatisztika | 63
|
| Schneider | 63
|
| 3. generációs | NSSP | Többszörös
összerendezések, neuronhálózat | 70
|
| LPAG | 68
|
| PHD | 73
|
| Konszenzus | JPRED | Több más módszer alapján
konszenzus | 73-75
|
| SOPMA
|
| CNRS
|
- 1. és 2. generációs módszerek: az egyes aminosavaknak a különböző
másodlagos szerkezetekben való előfordulásának gyakoriságai alapján
dolgoznak. Pontosság <70%, béta-szerkezetre
csak 28-48%, túl rövid hélixek és béta-szálak
- Chou-Fasman: Alfa-hélixet indít ott, ahol 6 egymás melletti
aminosav közül 4-nek 1,03 a hélixben levési valószínűsége, béta szálat ott,
ahol 5 egymás melletti aminosav közül 3-nak legalább 1,0 a bétában levési
valószínűsége. Ezután a hélixeket és béta-szálakat kiterjeszti mindkét
irányba addig, amíg 4 egymás melletti aminosav átlagos hélix-, ill.
béta-képzési valószínűsége 1,0 alá nem esik. Béta-kanyar jóslása hasonló
módon.
- GOR (Garnier-Osguthorpe-Robson): 17 aminosav szélességű ablakban
vizsgálja az aminosavak előfordulását, ezek alapján jósolja az ablak középső
pozíciójában lévő aminosavhoz tartozó másodlagos szerkezetet
- 3. generációs módszerek: a vizsgált szekvenciákhoz hasonlóakat
keresnek és ezekből többszörös összerendezést készítenek, majd a többszörös
összerendezésekben rejlő információt használják fel. A többszörös
összerendezések információt tartalmaznak az együtt mutálódó aminosavakról
(korrelált mutációk), ezek többnyire a térszerkezetben egymáshoz közel
vannak, tehát a többszörös összerendezés információt tartalmaz a fehérje
harmadlagos szerkezetéről, ily módon segíti a másodlagos szerkezet jóslását.
- Konszenzusos módszerek: több más módszert (2. és 3. generációs
módszereket) alkalmaznak, az eredmények konszenzusát veszik. Pl. JPRED: Példa
PHD módszer (ma a legjobb, akár 77% pontosság) [Burkhard Rost]
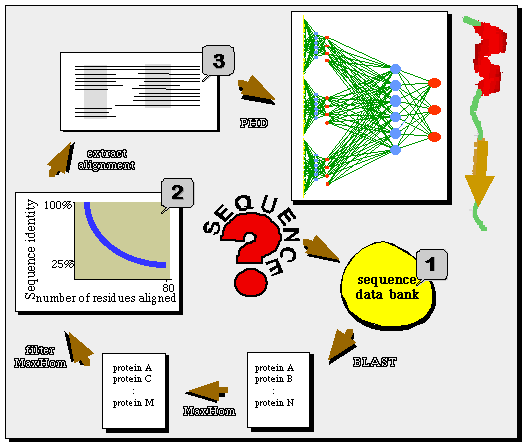
- adatbázisból kikeressük a rokon szekvenciákat (BLAST program)
- Elkészítjük ezek többszörös összerendezését (MaxHom program)
- Az összerendezés szűrése, jó homológok megtartása, újból összerendezés
- A végső összerendezés alapján mindegyik pozícióra elkészítjük az
előforduló aminosavcserék profilját
- Ez szolgál bemenetként a neuronhálózatnak
Bár a PHD módszer a legjobbnak látszik, mégsem mindig az, ezért helyes,
ha konszenzus módszert alkalmazunk, ill. több módszert együtt.
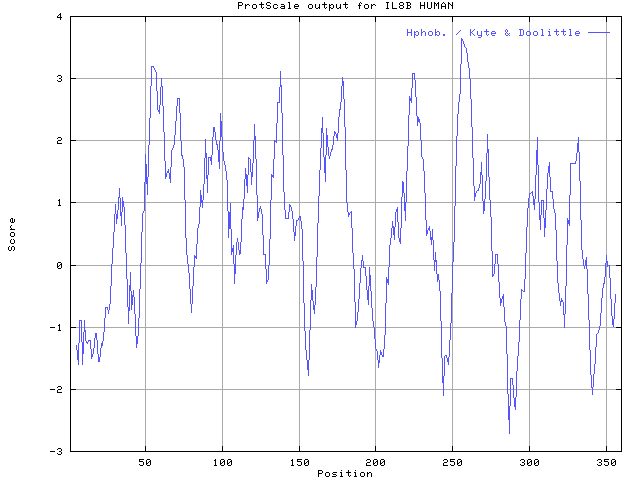
Oldószer általi hozzáférhetőség predikciója
- Felszíni vagy eltemetett az oldallánc
- Kezdetleges predikció: az oldallánc hidrofobicitása alapján -->
gyenge eredmény
- Jobb predikció: evolúciós információ bevitele többszörös
összerendezések útján --> 75% pontosságú jóslás (PHDacc)
Transzmembrán hélixek predikciója
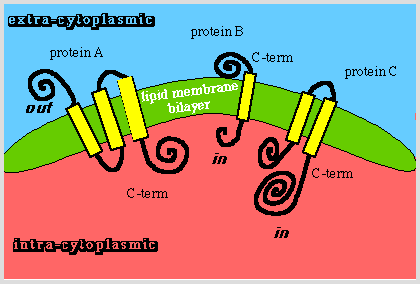
Transzmembrán fehérjék topológiája
2D predikciók
Oldallánc-kontaktusok predikciója
- Az összes kontaktusból elvben felépíthető a 3D szerkezet
- Próbálkoztak kontaktusokat jósolni a következők alapján:
- szekvenciában távoli aminosavak együtt előforduló
(korrelált) mutációi
- statisztika
- átlagtér-potenciálok
- neuronhálózatok
- Az eddigi eredmények nem kielégítőek
Fehérjeszerkezetek elemzése
Célja: a működés mechanizmusának részletes megértése, az egyes szerkezeti
motívumok felismerése, jelentőségük feltárása, stb.
Minőségellenőrzés
- PDB-ben lévő szerkezetek nagy része tökéletlen, hibákat tartalmaz. A
modellezéssel nyert szerkezetek még inkább.
- Minőségellenőrzés elvei:
- Kémiai felépítés: megvan-e minden atom, nem szakadt-e a lánc, stb.
- Sztereokémia: Kötéshosszak, kötésszögek, torziós szögek megfelelnek-e
az elvárható (a jó szerkezetek átlagából számított) értékeknek? Nincsenek-e
túl közel eső atomok?
- Fehérjetulajdonságok: Ramachandran-térképen a fi és pszi szögek
eloszlása (nincs-e valami a tiltott zónában, stb.); oldalláncok jó
környezetben vannak-e, megfelelően szoros-e a hidrofób mag pakoltsága, stb.
- Többféle program: PROCHECK,
WHAT_CHECK
- PROCHECK kimenetek pl.:
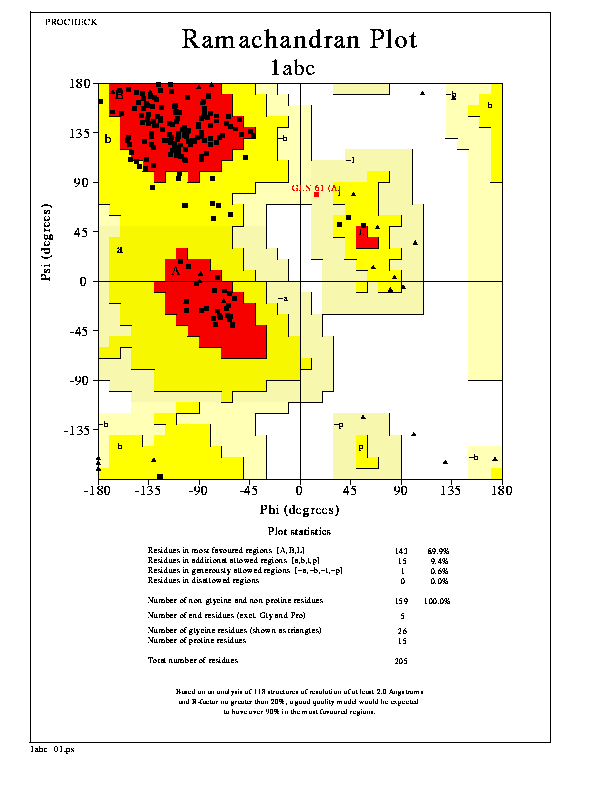
Ramachandran-plot
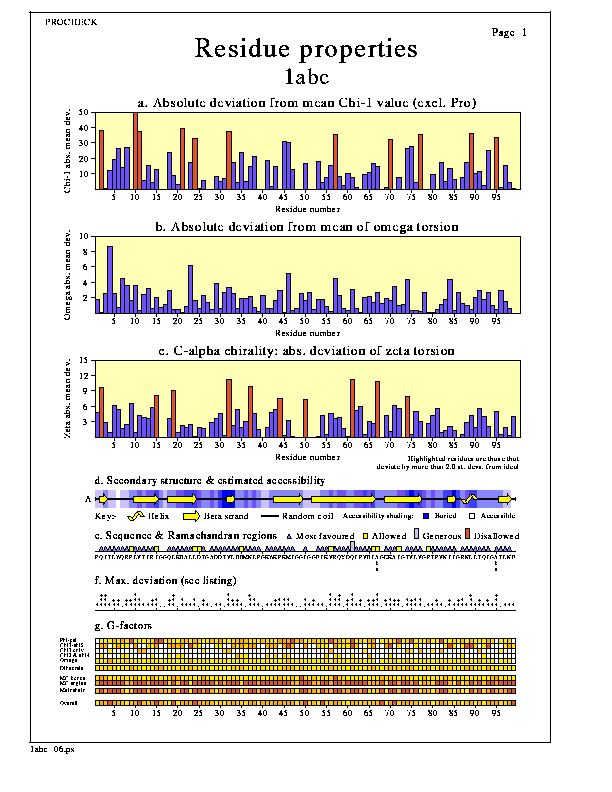
Oldallánc-paraméterek
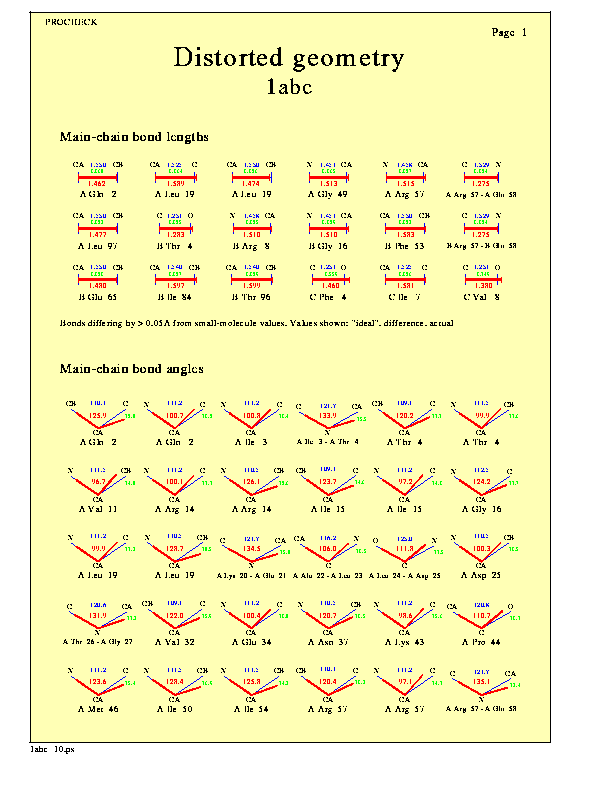
Geometriai torzulások
Másodlagos szerkezet
A meglévő atomi térszerkezet alapján hogyan definiáljuk a benne lévő
másodlagos szerkezeti elemeket?
- Másodlagos szerk. definiálható a fi, pszi szögek alapján is, de ez nem
megbízható
- Bevált: a hidrogénkötési mintázat alapján (pl. hélixek: i-->i+4
H-kötések, stb.)
- Standard módszer: DSSP program (és adatbázis). Példa
- Kódok: H: Hélix, E: Béta-szál, B: izolált H-kötés, S: görbület, G: 3-10
hélix, stb.
Szerkezeti motívumok
PROMOTIF
program és adatbázis.
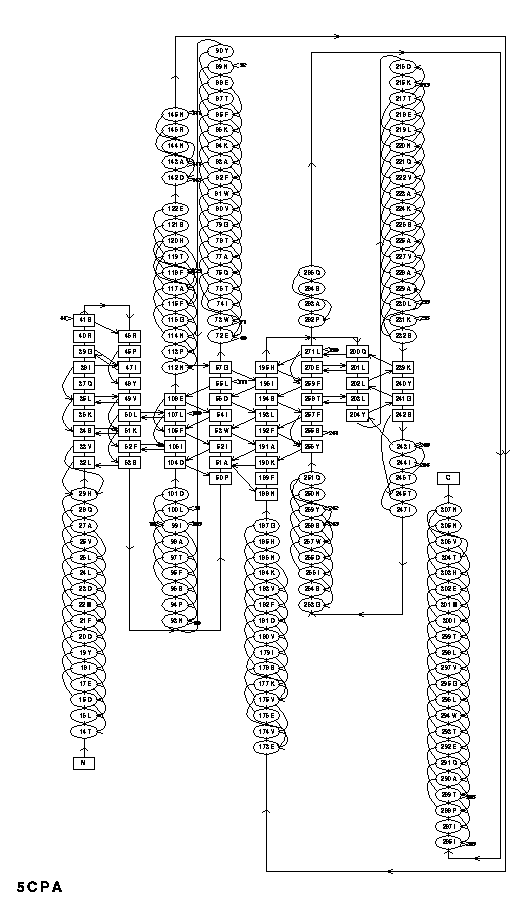
Főlánc H-kötései

Hélixek
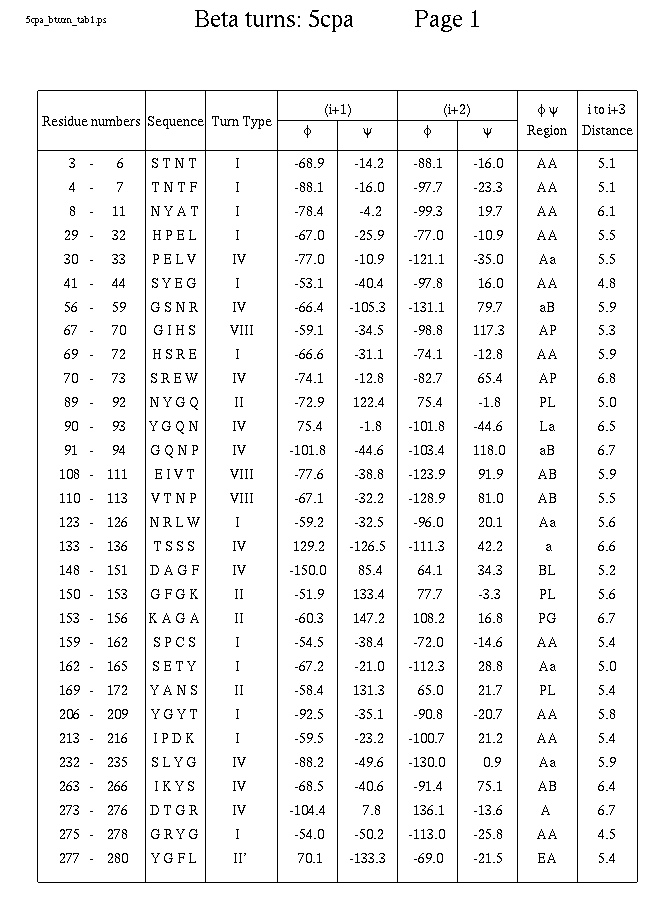
Béta turnök
Kölcsönhatás ligandumokkal
Vizualizálható: LIGPLOT
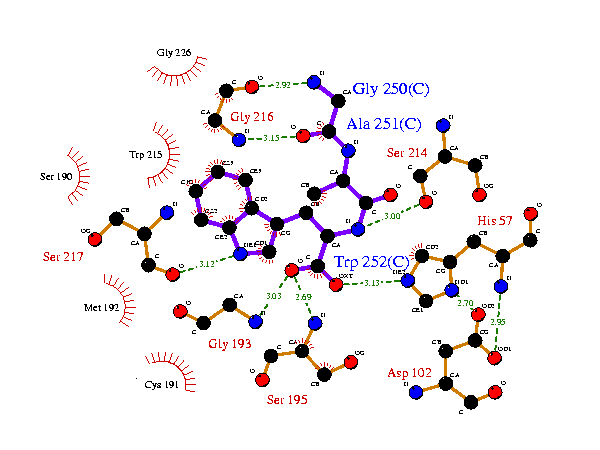
H-kötéseket és hidrofób felszíneket mutatja
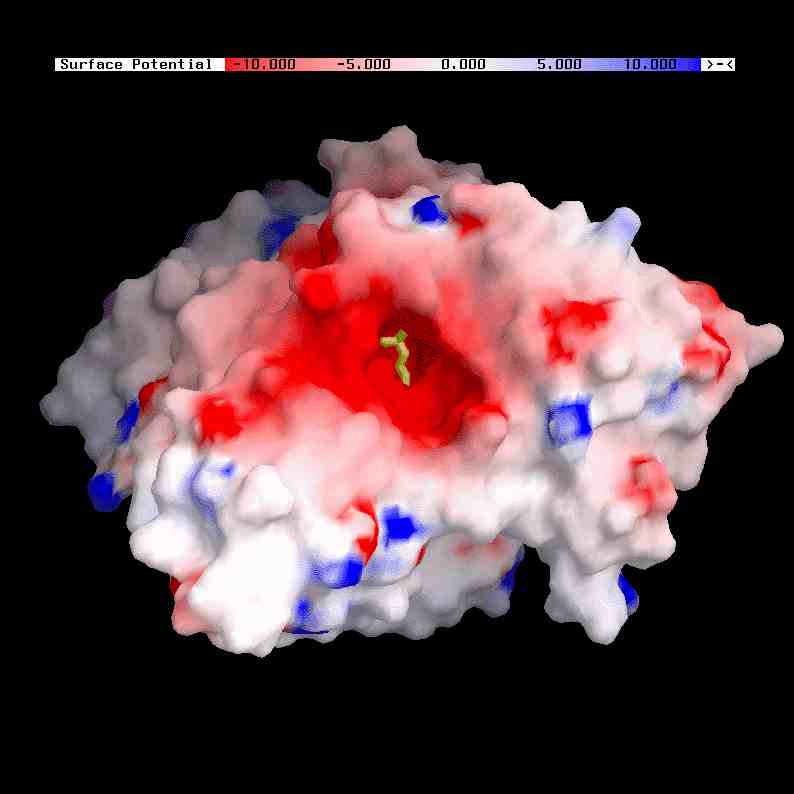
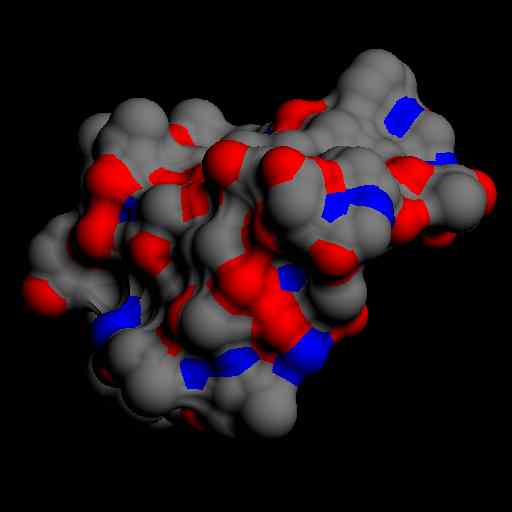
Töltésviszonyok
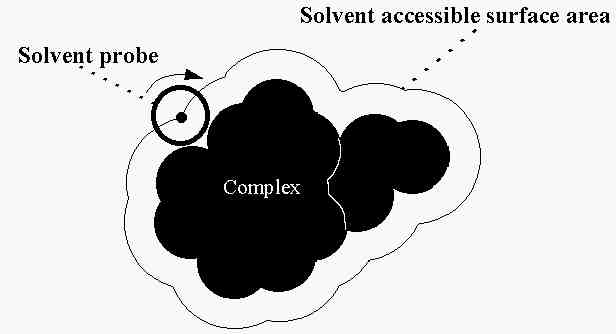
Felszínek, üregek