Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
6. Nukleotidszekvenciák analízise
- Bevezetés
- DNS-szekvenciák összeszerelése
- Génvadászati forgatókönyv
- A repetitív elemek kiszűrése
- Hasonlósági keresések
- ORF-detektálás
- Kodongyakoriság-eltolódások detektálása
- Funkcionális helyek detektálása
- Integrált génelemzés
- Webhelyek, példák
Bevezetés
Centrális dogma
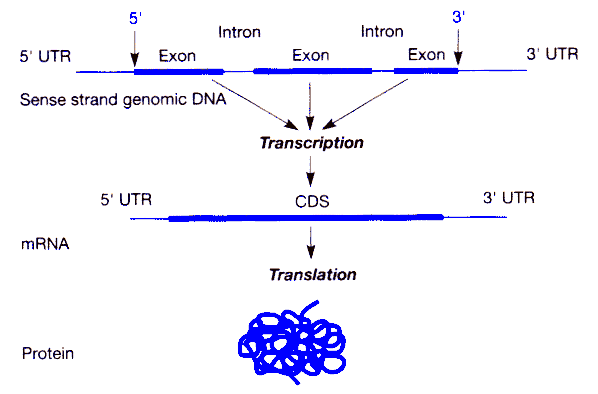
Rövidítések: UTR: Untranslated Region, CDS: Coding sequence
A genetikai kód
| |
T |
C |
A |
G |
|
| T |
Phe |
Ser |
Tyr |
Cys |
T |
| C |
| Leu |
Stop! |
Stop!/Sec |
A |
| Trp |
G |
| C |
Leu |
Pro |
His |
Arg |
T |
| C |
| Gln |
A |
| G |
| A |
Ile |
Thr |
Asn |
Ser |
T |
| C |
| Lys |
Arg |
A |
| Met/Start |
G |
| G |
Val |
Ala |
Asp |
Gly |
T |
| C |
| Glu |
A |
| G |
A genetikai kód közel univerzális (apróbb eltérések azért vannak egyes
mikroorganizmusokban, mitokondriumokban, stb.)
A TGA időnként nem Stop-ot, hanem szelenociszteint (Sec) kódol (21.
aminosav!)
DNS-szekvenciák összeszerelése
- cDNS szekvenálásánál 200-500 bázis hosszú darabokat kapunk (egy menetben
ekkorát lehet szekvenálni, újabban azért max. 1000-et is)
- Szekvenálási hibák: kb. 5% (hibás bázisok; kimaradt/tévesen beszúrt
bázisok: ún. fantom INDEL-ek)
- Emiatt mindkét szálat többször meg kell szekvenálni a megbízhatóság végett
- A teljes szekvenciát a darabokból kell összeszerelni és konszenzust
konstruálni:
AACCGTTTACGAAACCAGGTGC
AACCGTTTACGAAACCAGGTGCGCGCCCGCGGGAAT
AACCGTTTACGAACCCAGGTGC
(konszenzus:) AACCGTTTACGAAaCCAGGTGCGCGCGCGcGGGAATCCTAAAAA
CGCGCGCGCGGGAATCCTAAAAA
TGCGCGCGCGAGGGAATCCTAAAAA
|
Kisbetűk: kisebb megbízhatóság
- Összeszerelés: különféle programokkal, pl.
TIGR
Assembler
Génvadászati forgatókönyv
- Kiszűrni a repetitív elemeket (kiderül, hol nem lehetnek a
szabályozó és kódoló régiók
- Hasonlósági keresést végezni az ismert szekvenciák adatbázisain
- Megvizsgálni a kodongyakoriságok eltolódásait (a fehérjekódoló régiók
legegyértelműbb jele)
- Funkcionális helyeket (iniciáció, termináció, exon-intron határok,
promoterek, stb.) keresni az ismert szekvenciamintázatok alapján
- A fenti lépésekből nyert információt egységes képbe foglalni,
konszenzusos képet kialakítani
Fontos:
- A legtöbb program specifikus valamely fajra vagy élőlénycsoportra
- A programokat a megfelelő kontextusban kell használni (pl. exon-intron
határt ne keressünk cDNS-ben)
A repetitív elemek kiszűrése
- Repetitív elemek: rövid (vagy hosszabb) ismétlődő szekvenciadarabok
- Eukariota genomok jelentős részét teszik ki
- "Interspersed" repeat: a genomban elszórva találhatóak, többnyire
transzpozábilis elemek (pl. transzpozonok) inaktiválódott kópiái
- Számos családjuk van, adatbázisuk: RepBase
- Minden DNS-elemzés előtt ki kell szűrni ezeket a szekvenciából, mert
minden elemzőprogramot megzavarnának.
- Jelentősége: megmutatja, hol nem lehetnek szabályozó és kódoló
régiók
- Programok erre: pl. Censor, RepeatMasker: a RepBase
vagy hasonló adatbázis alapján szűrnek
- Pl. Censor futtatás eredménye:
Bemenő szekvencia: humán kreatin kináz génjének részlete
> HUMCKMM1
ggatccttcctccttggcctcccaaagtgctgggattacaggtgtgagccactgcacctg
gcctattacccttctcaggctctggagtccatccttctgctctgtctccctcagttcaat
tgttttttgttttttgttttttttttagacacagtctcgctctgtcaccaaggctggagt
gcagcagtgcgatcacagctcaccgcagcctcacctcccaggctcaagtgatcctcccat
ctcggcctctgagtagctgagactataggtgtgtccacatgtccggctaatttttgtatt
tttagtagagacagggtttcaccgcgttggccagggtggtcttgaactcctgagctcaag
caatcctcctgcctcagcctccttgttttgatttttagatcccacaaataacttgtgatg
tttgtctttctatacctggttcatttaacattttctttttcttttcttttcttttttttt
ttttttgtgagactgagtcttgctctgtcactcaggctggagggcaatggtgcatctcag
ctcactgcaacctccacctcctaggttcaagcaattcttatgcctcagcctcctggctag
ctgggattacaggcgtgtgtcaccatgccaggctaatttttgtacttttagtagagatgg
ggtttcaccatgttggccaggctggtcttgaactcctggcctcaagtgatccacccgcct
ccgcctctgcctcccaaagtgctgggattacgggcctgagccactgtgcccggcccatct
aacattttcactgtcaatcacaatgggattaaaactcctcccacagcccctagggacca
CENSOR futtatás eredménye:
Megtalált repetitív elemek:
kezdet vég elem neve
humckmm1 2 63 Alu-Jb 1 62 c
humckmm1 67 119 L1MA2 697 751 c
humckmm1 138 382 Alu-Jb 42 290 c
humckmm1 383 449 L1MA2 623 696 c
humckmm1 451 480 (TTTTC) 5 33 d
humckmm1 481 775 Alu-Sz 1 290 c
A repetitív elemektől megtisztított szekvencia (a törölt részek kiikszelve):
GXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXTATXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXTTGTTTTTTGTTTTTTGTXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXCATCTAACATTTTCACTGTCAATCACAATGGGATTAAAACTCCTCCCACAGCCCCTAGGGACCA
|
Hasonlósági keresések
A génazonosítás legjobb módszere, ha találunk a szekvenciaadatbázisokban
már meglévő, hasonló szekvenciát. Ennek módjai:
- A szekvencia összehasonlítása a GenBank-kal, ill. a dbEST-vel (BLASTN)
- A szekvencia mind a 6 lehetséges konceptuális transzlációjának
összehasonlítása a fehérjeszekvencia-adatbázisokkal (BLASTX)
- A szekvencia transzlációinak összehasonlítása a GenBank transzlált
változatával (TBLASTX)
- A szekvencia transzlációinak összehasonlítása másodlagos
fehérjeszekvencia-adatbázissal (pl. PROSITE)
Csapdák:
- Esetenként benne maradhat az mRNS-ben, így a cDNS-ben egy intron. Ekkor
ezt tévesen exonnak vélhetjük.
- A fehérjeszekvencia-adatbázisban nem valódi fehérjeszekvenciák is
lehetnek (szintén transzlációk)
- Az új szekvenciáknak csak kb. a fele mutat hasonlóságot valamilyen
régebbi szekvenciával
- Stb.
ORF-detektálás
- ORF: Open Reading Frame ("nyitott leolvasási keret"): olyan
nukleotidszekvencia-szakasz, melyet Stop kodon zár le.
- Melyik a valódi leolvasási keret? Ehhez előállítjuk mind a 6 lehetséges
transzlációt (a DNS mindkét szála leolvasható, egymással ellentétes
irányban, és mindkét szál háromféleképpen olvasható le), pl.
Your Nucleic Acid Sequence:
10 20 30 40 50
0 TCCATTGAGC CTTATACCAG TAACATCTAC ACTCGAAGAT CTTGTCAGGG
50 GAATTTCAGA TTGTGAATCC TCACTTACTG AAAGATCTTA CTGAGCGGGG
100 CTTGTGGAAT GAAGAGATGA AAAATCAGAT TATTGCATGC AATGGCTCCA
150 TTCAGTTTTC CTTTTTCAGA GCATACCAGA AATTCCTGAT GACCTGAAGC
200 AACTCTATAA GACCGTGTGG GAAATCTCTC AGAAGACTGT TCTCAAGATG
Six-Frame Amino Acid Translation:
Forward 0:
10 20 30 40 50
0 SIEPYTSNIY TRRSCQGNFR L!ILTY!KIL LSGACGMKR! KIRLLHAMAP
50 FSFPFSEHTR NS!!PEATL! DRVGNLSEDC SQD
Forward 1:
10 20 30 40 50
0 PLSLIPVTST LEDLVRGISD CESSLTERSY !AGLVE!RDE KSDYCMQWLH
50 SVFLFQSIPE IPDDLKQLYK TVWEISQKTV LKM
Forward 2:
10 20 30 40 50
0 H!ALYQ!HLH SKILSGEFQI VNPHLLKDLT ERGLWNEEMK NQIIACNGSI
50 QFSFFRAYQK FLMT!SNSIR PCGKSLRRLF SR
Reverse 0:
10 20 30 40 50
0 HLENSLLRDF PHGLIELLQV IRNFWYALKK EN!MEPLHAI I!FFISSFHK
50 PRSVRSFSK! GFTI!NSPDK IFECRCYWYK AQW
Reverse 1:
10 20 30 40 50
0 ILRTVF!EIS HTVL!SCFRS SGISGML!KR KTEWSHCMQ! SDFSSLHSTS
50 PAQ!DLSVSE DSQSEIPLTR SSSVDVTGIR LNG
Reverse 2:
10 20 30 40 50
0 S!EQSSERFP TRSYRVASGH QEFLVCSEKG KLNGAIACNN LIFHLFIPQA
50 PLSKIFQ!VR IHNLKFP!QD LRV!MLLV!G SM
|
- A valódi leolvasási keret valószínűleg a leghosszabb megszakítatlan szekvenciát
eredményező.
- A Start kodon azonosítása sokkal nehezebb (ATG a metionin kódja is), ld.
később.
Kodongyakoriság-eltolódások detektálása
- A nemkódoló és a kódoló régiókban az egyes kodonok (nukleotidhármasok)
gyakorisága jellegzetesen eltérő
- Különféle organizmusok más-más kodonpreferenciát mutatnak. Pl. a szerin
hatféle kodonjának gyakoriságai 5 fajban:
| Kodon | E. coli | D.
melanogaster | H.
sapiens | Z. mays | S. cerevisiae
|
|---|
| AGT | 3 | 1 | 10 | 3 | 5
|
|---|
| AGC | 20 | 23 | 34 | 30 | 4
|
|---|
| TCG | 4 | 17 | 9 | 22 | 1
|
|---|
| TCA | 2 | 2 | 5 | 4 | 6
|
|---|
| TCT | 34 | 9 | 13 | 4 | 52
|
|---|
| TCC | 37 | 48 | 28 | 37 | 33
|
|---|
- Organizmuson belül is változhat! Pl. E. coli-nál három kategória
- Sokféleképpen meg lehet ragadni, legsikeresebb: a hexamerek
(nukleotidhatosok) előfordulási gyakorisága
- Pl. GeneMark program valószínűséget számol (kódoló-e a régió) a
hexamergyakoriságok alapján, mind a 6 leolvasási keretben.
- Más programok (pl. GenScan) Markov-modellekkel dolgoznak:
Pozíciófüggő (inhomogén) ötödrendű Markov-modell: minden pozícióban
valamely nukleotid valószínűségét az előtte levő öt nukleotid milyensége
határozza meg, s ez a kerethez viszonyított pozíciótól is függ. A
valószínűségértékeket a már ismert gének alapján határozzák meg, új
szekvencia valószínűsége a modell alapján kiértékelhető.
Funkcionális helyek detektálása
Promoterek, exon-intron határok, transzlációiniciációs helyek,
terminációs helyek azonosítása, az ezekre jellemző szekvenciamintázatok
alapján
Az algoritmusok jóságának két paramétere:
- Érzékenység: a valóságos funkcionális helyek mekkora részét jósolja meg
helyesen
- Specificitás: a megjósolt helyek mekkora része valóságos
Különböző megközelítések:
- Konszenzusszekvencia alapján (a leggyakoribb nukleotidok megadása): nem
megbízható a variabilitás miatt
- Súlymátrix megadása: minden pozícióra közöljük a 4 nukleotid
gyakoriságát, ehhez hasonlítjuk az új szekvenciát. Korlátozott mértékben
sikeres.
- Bonyolultabb matematikai modellek (pl. rejtett Markov-modellek).
Ígéretes, de nem kiforrott.
Promoterek
- Számos program, az ismert promoterek szekvenciáinak könyvtára alapján
- Pl. TATA-box, "cap" szignál, stb.
- Kevéssé sikeres, mert pl. az emberi gének 30%-ánál nem találhatóak meg a
szokásos promoterszignálok
- A sikeresség nem nagyobb, mint a kodongyakoriság-eltolódások alapján
Poliadenilációs szignál
- A gének végén, ált. AATAAA konszenzusszekvenciával, melyet egy
bonyolultabb szignál követ
- A gének felénél hiányzik
- Nem megbízható
Transzlációs szignálok: start és stop kodon
Exon-intron határok
- Az exon-intron határokon (5' vég a donor hely, 3' vég az akceptor)
jellegzetes szekvenciák vannak, melyeket a spliceoszóma felismer
- Intronok szinte mindig GT-vel kezdődnek és AG-vel végződnek, ez kevés,
de segít leszűkíteni a lehetőségeket
- Bonyolultabb mintázatok súlymátrixa alapján elég hatásos detektálás, de
nem tökéletes
- Bonyolultabb matematikai modellek (pl. rejtett Markov) jelentős, de nem
drámai javulást eredményeznek
- Leghatásosabb: együttesen alkalmazni a kodongyakoriság-vizsgálattal (80%
fölé emelkedik az érzékenység)
Integrált génelemzés
- Az egyes predikciók hatékonyságát nagyon jelentősen növeli, ha a különböző
algoritmusokból származó információt együttesen vesszük figyelembe. Ez
vezetett az integrált génelemző programok kifejlesztéséhez
- A fenti eljárásokat egymás után alkalmazzák
- Számos ilyen program, szerver van, lásd pl.
Gene
Prediction Services, Gene
Recognition Programs
- 90% körüli érzékenységet, ill. specificitást lehet elérni velük.
Korlátaik
- Csak néhány organizmusra létezik ilyen program
- Néhány kivétellel csak akkor működnek jól, ha csak egyetlen gén van a
beadott szekvenciában
- Legtöbbször nagyon érzékenyek a szekvenálási hibákra
- Nem képesek kezelni az olyan eseteket, mint pl. alternatív splicing,
átfedő gének, különleges promoterek, stb.
- Ajánlott mindegyik programot használni (más-más algoritmusokkal
dolgoznak)
- Pl. a Genscan eredményessége egy 66 kilobázis méretű szakaszon:
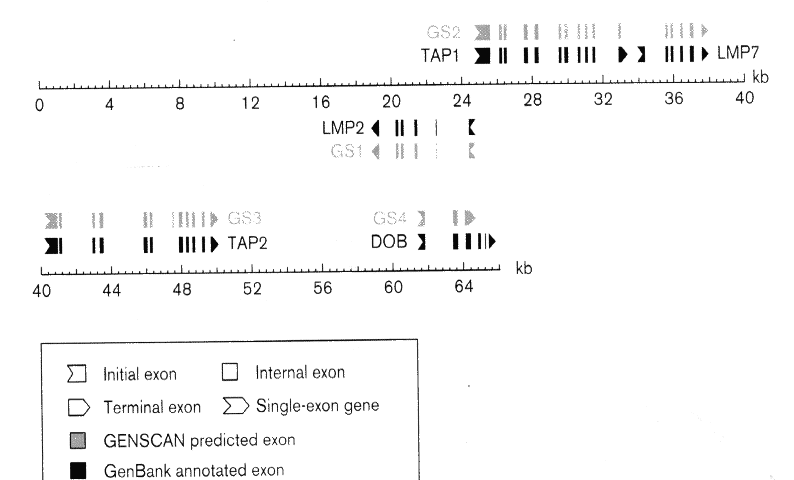
Fekete jelöli a valóságot, szürke a GenScan általi jóslást (ld.
jelmagyarázat). Az LMP2 gén jóslása tökéletes, de a TAP1-et és az LMP7-et a
GenScan egybeolvasztotta, a TAP2-be eggyel több exont jósolt, a DOB-nak
pedig túl korán véget vetett. Az exonok túlnyomó részét mégis hibátlanul
detektálta.
Webhelyek
Ld. pl.
Gene
Prediction Services, Gene
Recognition Programs