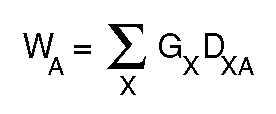Rejtett Markov-modellek (Hidden Markov Model, HMM): Olyan valószínűségi
modell, amely szekvenciákat "generál": tkp. lineáris lánc, amely egyezés
(Match, M), inszerció (I) és deléció (D) állapotokból áll, az ezek átmeneteit jellemző
számadatokkal. Pl.:
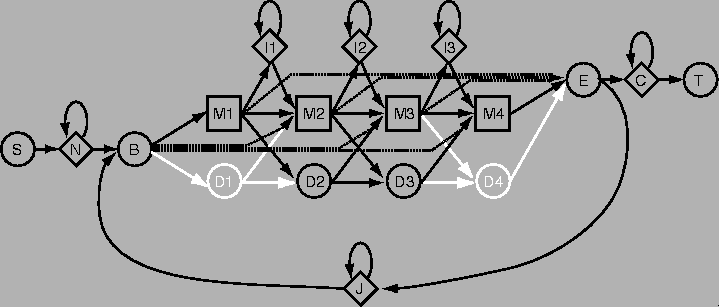
A rejtett Markov-modell (angolul hidden Markov model,
röviden HMM) egy képzeletbeli gép, amely szekvenciákat generál. A gépnek
véges sok állapota van, és ezek között lépked. Minden egyes állapotában vagy
minden egyes állapotváltáskor kibocsáthat egy szekvenciaelemet (tehát
aminosavat vagy nukleotidot), ezekből áll össze a gép által generált
szekvencia. A biológiai szekvenciák analíziséhez használt
HMM-eknek alapvetően háromféle állapota van: M (match, azaz egyezés), I
(insert, azaz inzerció) és D (delete, azaz deléció); ezeken kívül szokás
még kiinduló- és végállapotokat és egyéb speciális állapotokat is
definiálni. A fenti ábra egy HMM állapotdiagramját mutatja.
A körök és a négyzetek a gép állapotait, az összekötő nyilak az egyes
állapotok között lehetséges átmeneteket reprezentálják. Az M és az I
állapotok ún. "kibocsátó" állapotok, tehát amikor a gép ezekben az
állapotokban van, akkor kibocsát magából egy szekvenciaelemet (aminosavat
vagy nukleotidot). A D állapotok nem kibocsátó állapotok. Mindegyik M és I
állapothoz tartozik egy táblázat, amely megmondja, hogy az adott állapotban
a 20 aminosav, ill. a 4 nukleotid közül melyiket milyen valószínűséggel
bocsátja ki a gép (tehát a táblázat 20 vagy 4 számot tartalmaz). A HMM-nek
további paraméterei az egyes állapotok közötti átmenetek valószínűségei,
tehát az állapotdiagramon lévő, az egyes állapotokat összekötő nyilak
mindegyikéhez tartozik egy valószínűségérték. A HMM rendszerint annyi M, I
és D állapotot tartalmaz, amilyen hosszú szekvenciát tipikusan generál. A
fenti ábrán látható HMM például 4 M állapotot tartalmaz, tehát amennyiben
működése során nem megy át sem I, sem D állapoton és nem is kanyarodik
vissza a J állapoton keresztül, akkor 4 aminosavból vagy nukleotidból álló
szekvenciát generál.
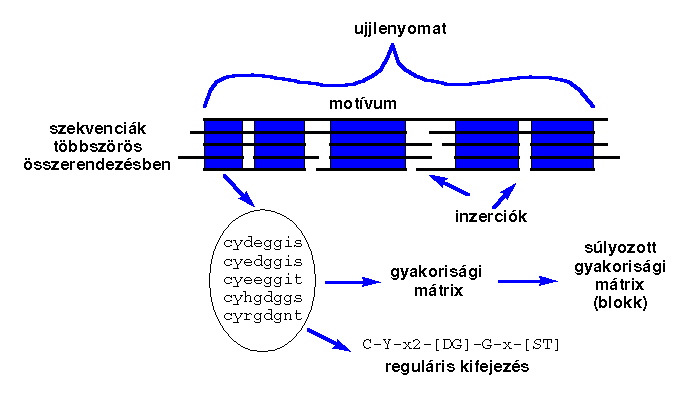
Ha van egy szekvenciahalmazunk, amely egymással rokon szekvenciákat
tartalmaz, akkor ennek a szekvenciahalmaznak az elemzésével, az egyes
pozíciókban lévő aminosavak gyakorisága és más mennyiségek alapján
definiálni lehet egy olyan HMM-et, amely olyan szekvenciákat generál, mint
amilyenek a kiinduló szekvenciahalmazban vannak. A HMM felépítése, az
állapotdiagram általában már eleve adott, a szekvenciahalmaz elemzésével
pedig meghatározhatjuk az M és az I állapotokban az egyes aminosavak, ill.
nukleotidok kibocsátásának valószínűségeit, valamint a gép egyes állapotai
közötti átmenetek valószínűségeit. A Pfam adatbázis ezeket a paramétereket
(pontosabban a számítások megkönnyítése végett a valószínűségek
logaritmusát) tartalmazza minden egyes fehérjecsaládra.
Ha tehát a rokon szekvenciákat tartalmazó halmaz alapján definiáltunk egy
HMM-et, akkor egy olyan modellünk van, amely ezt a bizonyos
szekvenciacsaládot jól leírja, és képes további olyan szekvenciákat
generálni, amelyek hasonlóak a kiinduló szekvenciahalmazban lévő
szekvenciákhoz. A szekvenciaanalízisnél azonban a HMM-nek nem ez a képessége
fontos, hanem egy másik tulajdonsága. Nevezetesen: a HMM segítségével meg
lehet határozni egy új szekvenciáról, hogy azt milyen valószínűséggel
generálhatja az adott HMM. Ha ez egy nagy valószínűségértéknek adódik, akkor
állíthatjuk, hogy a vizsgált, új szekvencia is beletartozik abba a
szekvenciacsaládba, amelyikből a HMM megkonstruálása során kiindultunk.
Hogyan határozható meg egy új szekvenciáról, hogy mekkora valószínűséggel
generálhatja azt egy bizonyos HMM? Egyszerű: meg kell nézni, hogy a HMM-nek
mely állapotok egymásutánján kell végigmennie (vagyis az állapotdiagramban
milyen útvonalat kell bejárnia) és mely állapotokban milyen szekvenciaelemet
kell kibocsátania ahhoz, hogy éppen a vizsgált (új) szekvencia jöjjön ki.
Ezután az állapotdiagramban bejárt útvonal mentén vett átmeneti
valószínűségeket és a kibocsátó állapotokban a megfelelő szekvenciaelem
kibocsátásának valószínűségeit összeszorozva adódik a vizsgált szekvencia
generálásának teljes valószínűsége. (Általában több útvonal is vezethet
ugyanahhoz a szekvenciához, ilyenkor vagy az összes lehetséges útvonal
összegét, vagy a legvalószínűbb útvonalat szokás figyelembe venni.)
A HMM-ek tehát olyan matematikai objektumok, amelyek
szekvenciamintázatokat képesek leírni (legyenek azok akár nagyméretű
fehérjeszekvenciák, akár néhány nukleotidból álló funkcionális helyek a
DNS-ben). Új szekvenciák esetében segítségükkel egzakt valószínűségérték
adható arra nézve, tartozhat-e az új szekvencia az adott HMM által leírt
mintázatú szekvenciák csoportjába.
A HMM-eket azért kedvelik, mert egzakt matematikai alapjuk van.
Ugyanakkor vannak hátrányaik is. Szerkezetükból adódóan nem képesek
megragadni például a szekvenciákban távolabb lévő aminosavak közötti
korrelációkat (pl. korrelált mutációk esetében). További hátrány, hogy egy
HMM által generált szekvencia egymástól független események egymásutánjának
az eredményeként áll elő, ami a valóságban nincs így: a szomszédos
szekvenciaelemekhez tartozó valószínűségértékek között a valóságban
általában összefüggés van. A fehérjeszekvenciákban például a hidrofób
aminosavak általában egymás mellé csoportosulnak, vagyis ha egy pozícióban
hidrofób aminosav generálódott, akkor a következő pozícióban nagyobb
valószínűséggel kellene szintén hidrofób aminosavnak generálódnia, mint
egyébként, de ezt a HMM nem képes figyelembe venni. E tökéletlenségek
ellenére a HMM-ek nagyon sikeresnek bizonyultak a szekvenciaanalízisben.
A "rejtett Markov-modell" kifejezésben a "rejtett" jelző egyébként arra
utal, hogy mi csak a modell működésének az eredményét, a kimenetet (azaz a
generált szekvenciát) ismerjük, maga a modell és a paraméterei számunkra
rejtettek. Tehát nekünk a kimenetből kell következtetnünk a modell
felépítésére és a működését leíró paraméterekre (az átmeneti és a
kibocsátási valószínűségekre).
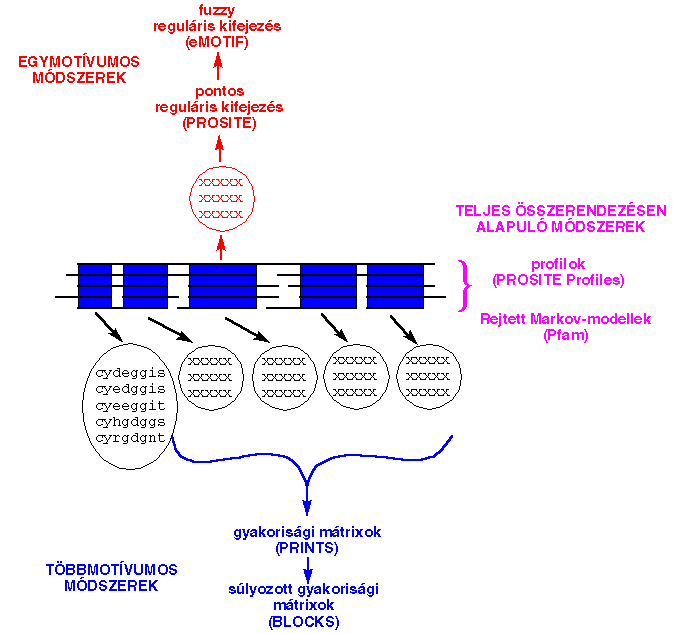
PROSITE-ban és Pfam-ban.