Ez a tananyag 2001-ben készült, így sok tekintetben elavult.
1. Mi a bioinformatika?
Vázlat: Ez a kurzus. Mi a bioinformatika? Bioinformatika a
szakirodalomban. A bioinformatika céljai. A biológiai információ típusai és
elemzési módszerei. Az adatok csoportosítása hasonlóságok alapján. A
bioinformatikai "spektrum". A szekvencia/szerkezet deficit. Genomprojektek.
Miért fontos a bioinformatika? Szekvenciaanalízis. Az ördög a részletekben
lakozik. Pár jótanács. Bioinformatikai webhelyek.
Ez a kurzus
- Anyag a weben: https://bioinformatika.szialab.org/
- Felépítés (kb.):
- Mi a bioinformatika?
- Adatbázisok
- Páronkénti szekvencia-összerendezés
- Többszörös szekvencia-összerendezés, filogenetikai analízis
- Másodlagos adatbázisok
- Nukleotidszekvenciák analízise
- Fehérjeszekvenciák analízise
- Fehérjeszerkezetek elemzése
- Fehérjék térszerkezetének jóslása
- Genomika
- Bioinformatikai stratégiák. Bioinformatikai programcsomagok.
- Fontos fogalmak bemutatása, nem a konkrét eszközökre koncentrálunk (ezek
változnak)
- Előadás utolsó része: gyakorlati példák
- Irodalom:
Mi a bioinformatika?
Bioinformatika: számítógépes módszerek kidolgozása és
alkalmazása a biológiai információ kezelésére és elemzésére.
- A szót az 1980-as évek közepén találták ki, legtágabb értelemben: minden,
amit számítógéppel csinálnak, és köze van a biológiához.
- Szűkebb értelemben: a biológiai szekvenciaadatok kezelése és
elemzése, ill. a 3D szerkezeti információ kezelése és elemzése.
Egy újabb definíció az Oxford English Dictionaryból:
Bioinformatics is conceptualizing biology in terms of
molecules (in the sense of physical-chemistry) and applying "informatics
techniques" (derived from disciplines such as applied maths, computer
science and statistics) to understand and organise the information
associated with these molecules, on a large scale. In short, bioinformatics
is a management information system for molecular biology and has many
practical applications.
(Hevenyészett fordításban) A bioinformatika a biológia fogalmi
megragadása a (fizikai-kémiai értelemben vett) molekulák segítségével, és
(az alkalmazott matematikából, a számítógéptudományból, a statisztikából és
más tudományágakból származó) "informatikai módszerek" alkalmazása az
ezekkel a molekulákkal kapcsolatos információ megértésére és
megszervezésére, nagy léptékben. Röviden, a bioinformatika egy
információmenedzselési rendszer a molekuláris biológia számára, és sok
gyakorlati alkalmazása van.
Bioinformatika a szakirodalomban
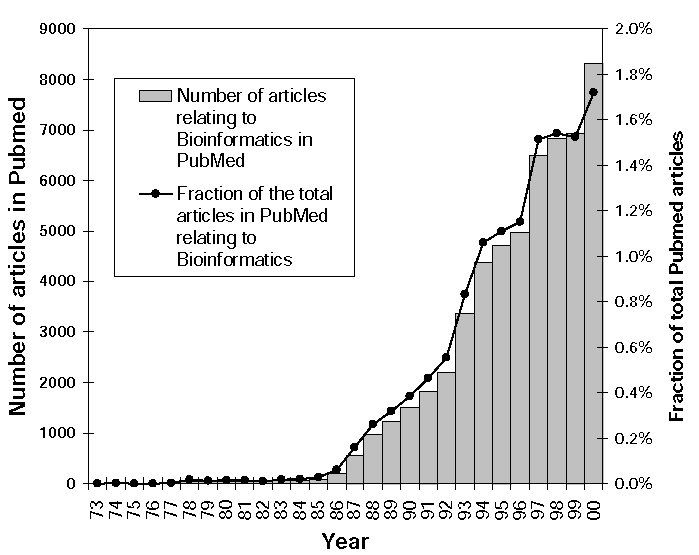
A bioinformatikai témájú cikkek aránya nő, mára kb. 2%. Egyes vélemények
szerint lassan már nem lesz szükség kísérletekre, mindent szimulálni fogunk.
A bioinformatika céljai
- Adatbázisok létrehozása és karbantartása.
Az adatok megszervezése, rendezése oly módon, hogy a kutatók könnyedén
hozzáférhessenek a meglévő információhoz, és hozzátehessenek újat.
- Eszközök, módszerek kifejlesztése az adatok elemzésére. Az adatok
haszontalanok, amíg nem elemeztük őket.
- Az eszközök és módszerek alkalmazása az adatok elemzésére, és az
eredmények értelmezése a biológia szempontjából.
A biológiai információ típusai és elemzési módszerei
| Az adatok forrása | Az adathalmaz mérete | Bioinformatikai témák
|
|---|
| Nyers DNS-szekvenciák | 12 millió szekvencia, 13 milliárd
bázis | - A kódoló és nem-kódoló régiók elkülönítése
- Az intronok és exonok
azonosítása
- A géntermékek predikciója
- Igazságügyi elemzések
|
| Fehérjeszekvenciák | 400 000 szekvencia (egyenként kb. 300
aminosav) | - Szekvenciaösszehasonlítási algoritmusok
- Többszörös
szekvenciaillesztő algoritmusok
- Konzerválódott szekvenciamotívumok
azonosítása
|
| Makromolekuláris szerkezetek | 15 000 szerkezet (egyenként kb.
1000 atom koordinátái) | - Másodlagos és harmadlagos szerkezet
jóslása
- 3D szerkezeteket illesztő algoritmusok
- Fehérjegeometriai
mérések
- Felszín, térfogat és alak számítása
- Intermolekuláris
kölcsönhatások
- Molekulaszimulációk (energiafüggvény, molekuláris
mozgások, dokkolás)
|
| Genomok | 300 teljes genom (egyenként 1,6 millió--3 milliárd
bázis) | - Az ismétlődések jellemzése
- Szerkezetek hozzárendelése
génekhez
- Filogenetikai analízis
- Genomi méretű felmérések
(fehérjetartalom jellemzése, anyagcsere-útvonalak)
- Kapcsoltság elemzése
egyes betegségek és gének összefüggésének vizsgálatához
|
| Génexpressziós adatok | legnagyobb: kb. 20 időpont az élesztő kb.
6000 génjénél | - Az expressziós mintázatok korrelációjának
vizsgálata
- Az expressziós adatok összekapcsolása a szekvencia-,
szerkezeti és biokémiai adatokkal
|
| Egyéb: Szakirodalom | 11 millió szakcikk | - Elektronikus
könyvtárak az automatizált irodalomkutatáshoz
- Tudásadatbázisok irodalmi
adatokból
|
| Egyéb:
Anyagcsere-útvonalak | | - Reakcióútvonal-szimulációk
|
Az adatok csoportosítása hasonlóságok alapján
Fontos: Az információ nagy része csoportokba rendezhető, értelmes biológiai
hasonlóságok alapján. Ez számos bioinformatikai módszer alapja. Pl.:
- a genomban sok ismétlődő szekvenciarészlet van
- a gének csoportosíthatóak funkciójuk (pl. enzimhatás) szerint vagy az
anyagcsere-útvonalak szerint
- különböző fehérjéknek gyakran hasonló a szekvenciájuk
- véges számú különböző alapvető fehérjeszerkezet létezik (becslések 1000
és 10 000 közé teszik)
--> "véges alkatrészlista" (az élőlények véges számú alkatrészből
épülnek fel)
Mintázatfelismerés és predikció
Két alapvető művelet a bioinformatikában
- Mintázatfelismerés: a hasonlóságok megtalálása
- A már ismert, hasonló funkciójú/szerkezetű fehérjéket megvizsgálva megkeresünk
valamely, a funkcióra/szerkezetre jellemző, konzerválódott sajátosságot
- Ezt használjuk fel új szekvenciák funkciójának/szerkezetének
azonosítására
- Feltétel: az új szekvencia olyan fehérjéhez tartozzon, amihez hasonlót már
"láttunk"
- Predikció:
- A funkció vagy a térszerkezet megjóslása, hasonlóság alapján vagy
másképpen
- A bioinformatika "Szent Grálja": a szekvenciából megjósolni a
térszerkezetet
MFENITAAPADPILGLADLFRADERPGKINLGIGVYKDETGKTPVLTSVKKAEQYLLENETTKNYLGIDGIPEFG
RCTQELLFGKGSALINDKRARTAQTPGGTGALRVAADFLAKNTSVKRVWVSNPSWPNHKSVFNSAGLEVREYAYY
DAENHTLDFDALINSLNEAQAGDVVLFHGCCHNPTGIDPTLEQWQTLAQLSVEKGWLPLFDFAYQGFARGLEEDA
EGLRAFAAMHKELIVASSYSKNFGLYNERVGACTLVAADSETVDRAFSQMKAAIRANYSNPPAHGASVVATILSN
DALRAIWEQELTDMRQRIQRMRQLFVNTLQEKGANRDFSFIIKQNGMFSFSGLTKEQVLRLREEFGVYAVASGRV
NVAGMTPDNMAPLCEAIVAVL
==>
- A felgombolyodás problémája: az aminosavsorrend meghatározza a
térszerkezetet (Anfinsen-kísérlet óta tudjuk), de még ma sem értjük, hogyan
[chaperonok nem számítanak]
- Csak másodlagosszerkezet-jóslás megy, korlátozott megbízhatósággal
- Várhatóan így marad még pár évtizedig
Homológia és analógia
Homológia = közös evolúciós eredet (szekvenciák esetében).
Nincs mértéke (nem fejezhető ki %-ban!!!), két szekvencia vagy homológ, vagy
nem.
Analógia: hasonlóság, közös evolúciós eredet nélkül
Két fehérje analóg lehet, ha:
- szerkezetük hasonló, de nincs közöttük szekvenciális hasonlóság, vagy
- azonosak a katalitikus csoportjaik, de térszerkezetük különböző
Ilyenkor közös evolúciós eredetre nem lehet következtetni. Inkább
konvergens evolúció állhat a háttérben.
Pl. szubtilizin és kimotripszin: mindkettő szerin proteáz, a His/Asp/Ser
katalitikus triáddal, de térszerkezetük teljesen eltérő:
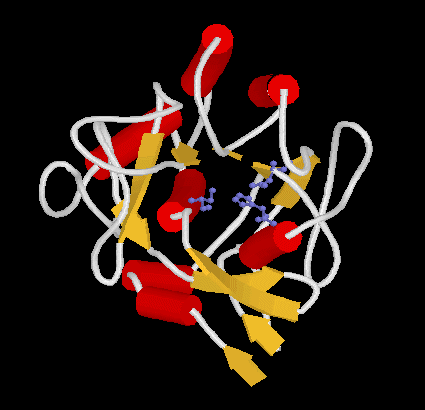
Szubtilizin
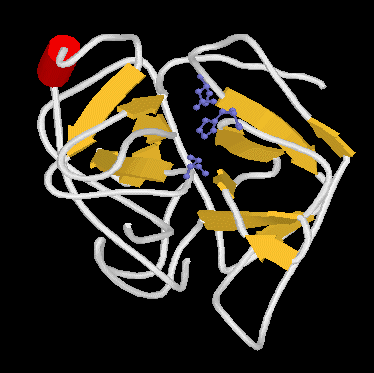
Kimotripszin
Ortológia és paralógia
A homológia két típusa
- Ortológia: két gén ortológ, ha két különböző fajban találhatóak,
és egy közös ős-génből származnak, mely a két faj közös ősében volt jelen. Ugyanazt a funkciót
szolgálják, a két fajban. Példa: laktát dehidrogenáz (ill. génje) az emberben és az
egérben.
- Paralógia: két gén paralóg, ha ugyanabban az organizmusban
találhatóak, és egy közös ős-génből génduplikáció és azt követő divergens
evolúció útján alakultak ki. Többnyire különböző, de egymással
összefüggésben lévő funkciójuk van. Példa: a
hisztidin-bioszintézis enzimei (ill. ezek génjei) emberben (nagyon hasonló szerkezetűek, de
más-más reakciót katalizálnak).
A bioinformatikai "spektrum"

Bioinformatikai módszerek két dimenziója:
- mélység: (függőleges tengely): fizika
veszünk egy fehérjét, és azt igyekszünk minél mélyebben megérteni.
Génszekvencia-fehérjeszekvencia-térszerkezet-geometria-kötőhelyek-gyógyszertervezés
- szélesség: (vízszintes tengely) informatika
a gén összehasonlítása más
génekkel. Páronkénti, majd többszörös szekvenciaillesztés,
szekvenciamintázatok azonosítása, filogenetikai elemzés, teljes genom
elemzése, stb.
A szekvencia/szerkezet deficit
Az ismert fehérjeszekvenciák és az ismert térszerkezetek számának
növekedése az utóbbi években:
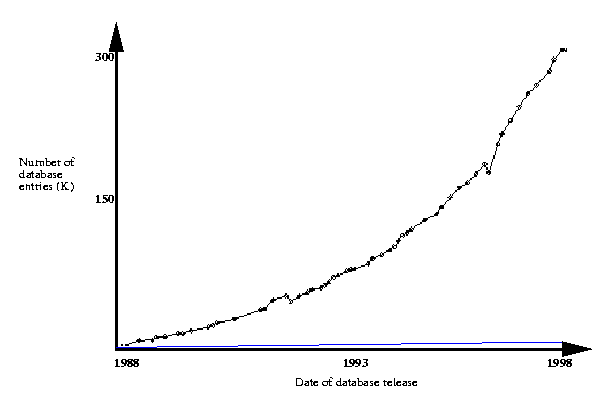
- Mintegy 200-szor több a szekvencia, évente megduplázódik.
- 1998: kb. 300 000 ismert szekvencia (sőt, az ún. EST adatbázisokat is
figyelembe véve milliók), kb. 1500 egyedi térszerkezet
- A helyzet a genomprogramokkal csak rosszabbodik
- Percenként egy új szekvencia. A későbbi nagy szerkezetmeghatározó
projektek is max. napi 5 új szerkezetet szolgáltatnak majd.
- Nagy információs deficit, itt van szerepe a bioinformatikának.
Genomprojektek
Már megszekvenált genomok (ld. http://www.ebi.ac.uk/genomes/:
- Eukarioták:
- Élesztő
- Caenorhabditis elegans (féreg)
- Drosophila melanogaster (muslica)
- Arabidopsis thaliana (lúdfű)
- Ember
- Egyéb:
- kb. 50 baktérium
- 11 archeon
- >100 organellum
- több száz vírus és fág
Folyamatban lévő fontos genomszekvenálási projektek
- ember
- egér
- kutya
- patkány
- stb.
A humán genom projekt
- Hivatalosan 1990-ben indult, az USA Dept. of Energy és National
Institutes of Health irányításával
- Tervezett befejezés: 2003
- Célok:
- azonosítani az emberi DNS mintegy 100 000 (újabb infók szerint csak
30 000) génjét
- meghatározni a kb. 3 milliárd bázis pontos sorrendjét
- ezt adatbázisokban tárolni
- fizikai géntérképet készíteni
- meghatározni a szekvencia variációit (polimorfizmusok)
- modellorganizmusok genomját szekvenálni (muslica, egér)
- gyorsabb, hatékonyabb szekvenáló módszereket kifejleszteni
- új adatelemző módszereket kifejleszteni
- a projekt etikai, jogi és társadalmi vonatkozásaival foglalkozni
- 1996-ig: térképezés, ezután szekvenálás
- Állami projekt: a kromoszómákat egyre kisebb szakaszokra darabolva, a
darabokat vektorokba klónozva úgy szekvenálnak, hogy mindig tudják, melyik
darabot vizsgálják.
- 1998: Craig Venter céget alapít: teljes genom shotgun szekvenálását
alkalmazva gyorsabban és olcsóbban akar elkészülni, mint az állami projekt
- Shotgun módszer: a genomot átfedő módon apró darabokra tördelve a
darabokat megszekvenáljuk, majd az átfedések alapján számítógéppel
összerakjuk. Teljes genom esetén (térkép nélkül) kétséges a repetitív
szekvenciák miatt.
- 1999. december: 22. kromoszóma készen van
- 2000. május: 21. kromoszóma
- 2000. június: durva vázlat (90% lefedve, töredékesen)
- 2001. február: a durva vázlat (draft) teljesen kész
- Ma: a genomszekvencia kb. 60%-a véglegesített, a többi csak vázlat
(darabkák orientációja és sorrendje bizonytalan, hibaarány nagy)
Miért fontos a bioinformatika?
- Genomprojektek --> Óriási tömegű szekvenciaadat: megfejtetlen kód
- Bioinformatika feladata: a szekvenciákban kódolt információ megfejtése:
- térszerkezetek
- funkciók
- evolúciós összefüggések
- funkció és evolúciós összefüggések térszerkezetből is
Szekvenciaanalízis
Az ördög a részletekben lakozik
A szekvenciaanalízis során számos csapdát kell elkerülni:
- Az ortológia és a paralógia bonyolult viszonyai
új szekvenciák vizsgálatánál megnehezítik annak eldöntését, hogy a
funkcionális információ mennyire vihető át az új fehérjére (a talált hasonló
szekvencia lehet az ortológ paralógja egy másik organizmusban). Emiatt az automatikus
funkcióhozzárendelés sok esetben hibás (és ezt adatbázisokban is megtalálni)!
- A szekvenciahasonlóság sokszor csak a szekvencia egy részére vonatkozik,
pl. moduláris fehérjék esetében.
Moduláris fehérjék
- Modulok: olyan fehérjedomének, amelyek cserélhető építőkövekként
szerepelnek
- Nem (csak) génduplikációval, hanem shuffling révén is terjednek
- Pl.:
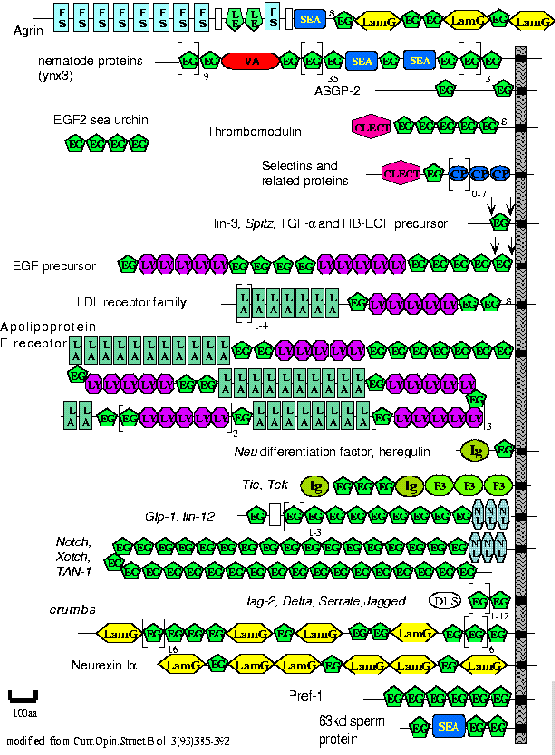
- funkciójuk változhat attól függően, milyen fehérje részét képezik
- Az evolúció "bütyköl", változatosan használja fel a modulokat. Hasonlat: Rube Goldberg rajzai (a biológia
metaforája):
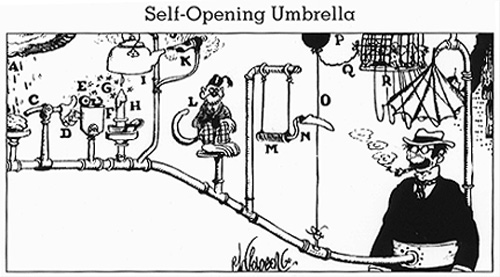 Az esőcseppek (A) ráhullanak az aszalt szilvára (B), mely megduzzad
és meglöki a kart (D), melynek másik végén a műkéz (D) megforgatja a kiürült
öngyújtó (F) dörzskerekét. A tűzkőből (E) kirepülő szikrák (G) meggyújtják a
gyertyát (H), mely felforralja a kannában (I) lévő vizet. A kilépő gőz (J)
megfújja a sípot (K). A cirkuszi majom (L) azt hiszi, hogy ez a jel az
attrakció megkezdésére, és átugrik a trapézra (M), mely lengés közben a kés
(N) élével elvágja a zsinórt (O), ezzel elengedve a léggömböt (P). A léggömb
felszáll, a hozzá kötözött madzag (Q) kinyitja a kalitka ajtaját, s a
madarak (R) kirepülnek, zsinórokkal felemelve az esernyő pálcáit.
Az esőcseppek (A) ráhullanak az aszalt szilvára (B), mely megduzzad
és meglöki a kart (D), melynek másik végén a műkéz (D) megforgatja a kiürült
öngyújtó (F) dörzskerekét. A tűzkőből (E) kirepülő szikrák (G) meggyújtják a
gyertyát (H), mely felforralja a kannában (I) lévő vizet. A kilépő gőz (J)
megfújja a sípot (K). A cirkuszi majom (L) azt hiszi, hogy ez a jel az
attrakció megkezdésére, és átugrik a trapézra (M), mely lengés közben a kés
(N) élével elvágja a zsinórt (O), ezzel elengedve a léggömböt (P). A léggömb
felszáll, a hozzá kötözött madzag (Q) kinyitja a kalitka ajtaját, s a
madarak (R) kirepülnek, zsinórokkal felemelve az esernyő pálcáit.
- Sok ilyen rajzon szerepel majom, ha csak azt látjuk, nem tudunk a többi
elemre, sem az egész rendszer funkciójára következtetni.
- Ha a majom hiányzik, nehezen tudjuk megjósolni, mi lehet a helyén, túl
sok lehetőség van
- A biológiai rendszerek is így épülnek fel részekből.
- Az új szekvenciák kb. egyharmadának egyáltalán nem lehet a funkciójára
következtetni az ismert funkciójú fehérjék szekvenciái alapján, mert a
funkcionális jellemzés nem képes lépést tartani a szekvenciaadatbázisok
növekedésével
- Még a nagy szekvencia- és szerkezeti hasonlóság sem jelent mindig
funkcionális azonosságot vagy rokonságot. Példa: alfa-laktalbumin és
lizozim: 50% szekvenciaazonosság, lényegében azonos térszerkezet, teljesen
más funkció (lizozim baktérium sejtfalát emészti, alfa-laktalbumin a laktóz
szintáz szabályozófehérjéje)

Alfa-laktalbumin

Lizozim
A szekvencia és a szerkezet szerepe a funkció meghatározásában
- A szerkezet a váz, amelyre a szekvencia fel van fűzve. A funkció
megállapításához szükséges a szekvencia részleteinek ismerete. Analógia: szoba
és berendezése (csak a berendezés részleteiből tudhatjuk meg, mire való a
szoba).
- A funkció megállapításához a biológiai kontextust is ismerni kell
(milyen épületben van a szoba)
A bioinformatikai programok, eljárások, algoritmusok nem végleges,
biztos válaszokat adnak, csak segítenek leszűkíteni a lehetőségek körét és
kísérleteket tervezni a kérdések eldöntésére. A valódi válaszokat a
biológiai háttérismeretek fényében találhatjuk meg.
Pár jótanács
Ne higgyük el mindig:
- ami az adatbázisokban van (félrevezető vagy hibás lehet a szekvencia
vagy a funkcióhozzárendelés)
- amit a programok mondanak (a programban hiba lehet, az eredmény
félrevezető lehet)
- amit a webszerverek mondanak (dettó)
- amit a szakirodalomban olvasunk (a tévedések nem ritkák)
Továbbá:
- Ne ragadjunk le egy "legjobbnak vélt" módszernél, adatbázisnál. Ne
ragadjunk le a részleteknél. Sokféle megközelítést alkalmazva keressünk
konszenzusos képet.
Bioinformatikai webhelyek
EMBL, SRS
NCBI (Medline, Genbank, stb.)
Expasy, Swissprot, Amos
CCP11 projekt